MoMKD: The Momentum Memory That Teaches Cancer Histology to Think Genetically
How a team at Wake Forest University School of Medicine built a cross-modal distillation framework that transfers the predictive power of expensive genomic assays into histology-only models—without ever requiring genomics at inference time.
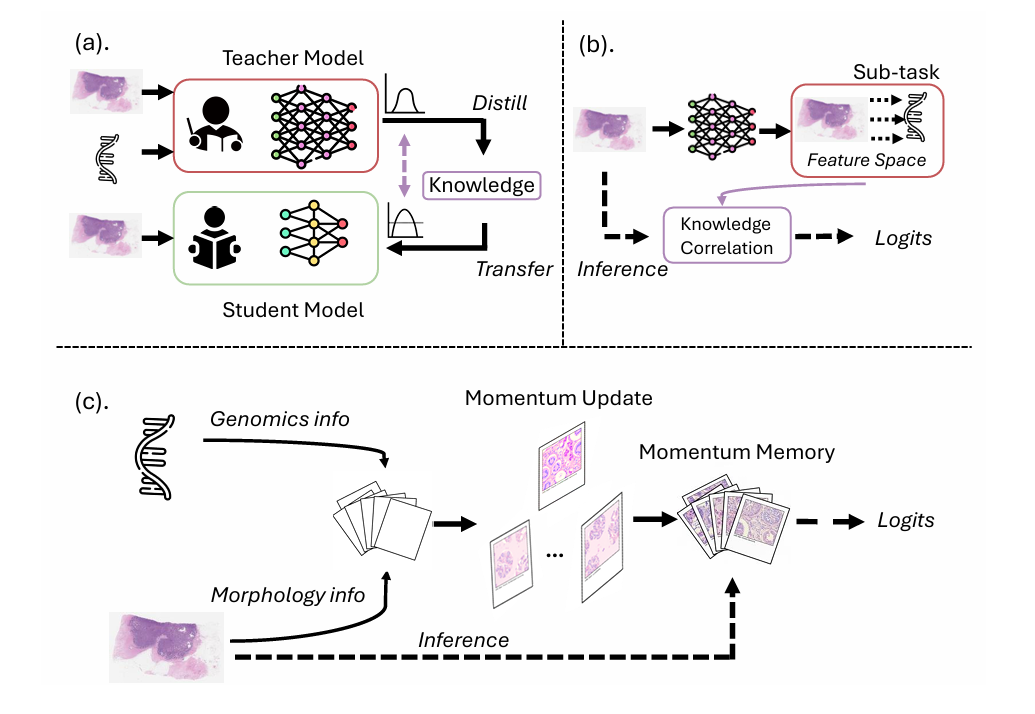
There is a fundamental tension sitting at the heart of computational pathology. The most clinically meaningful biomarkers in cancer—molecular subtypes, gene-expression signatures, recurrence risk scores—are defined not by what a tumor looks like under a microscope, but by what is happening at the genomic level. Yet genomic assays are slow, expensive, and unavailable in resource-limited settings. Pathology slides, by contrast, are collected routinely for nearly every cancer patient. If a model could learn to read the molecular story hidden within those slides, the clinical and economic implications would be enormous.
This is the motivation behind MoMKD—Momentum Memory for Knowledge Distillation—developed by researchers at Wake Forest University School of Medicine. The framework takes a practical and carefully engineered approach: train a multimodal model that ingests both histopathology images and genomic data, then use knowledge distillation to compress everything the genomics branch learned into the histology branch alone. At inference time, you only need the slide. The genomics knowledge is already baked in.
What makes MoMKD different from previous distillation attempts is how it handles the fundamental instability of cross-modal alignment. Rather than forcing both modalities to directly match each other within each training mini-batch—an approach that is inherently fragile and limited by sample diversity—MoMKD introduces a slowly-evolving momentum memory: a compact, class-conditioned dictionary that accumulates genomic and histopathology statistics across the entire training trajectory. Both modalities align to this shared semantic anchor instead of to each other, turning an unstable direct-matching problem into a stable dictionary-lookup problem.
Evaluated across three biomarker prediction tasks on the TCGA-BRCA benchmark and an independent in-house clinical cohort, MoMKD consistently outperforms both WSI-only MIL baselines and previous multimodal distillation methods—sometimes by wide margins. The results make a compelling case that the right memory architecture is not a peripheral design detail. It is the central ingredient that makes cross-modal distillation work.
Why Batch-Local Distillation Has Always Been the Wrong Framing
To understand what MoMKD solves, it helps to first understand what existing knowledge distillation methods in pathology actually do—and where they break down.
The standard paradigm in cross-modal KD for pathology treats the distillation process as a feature-matching problem within each mini-batch. Either both modalities are pushed into the same latent space so that corresponding pairs sit close together, or one modality is used to construct auxiliary regression targets that the other modality learns to predict. Both strategies share the same structural weakness: their supervisory signal is defined entirely by whatever samples happen to be present in the current mini-batch.
In practice, this fragility is severe. Mini-batches in pathology contain gigapixel whole slide images, where background regions dominate and meaningful tumor patches are sparse. The number of distinct classes represented in any given batch is limited, which means the contrast between positive and negative examples is thin. And the geometries of genomic embeddings and histopathology embeddings are fundamentally asymmetric—global transcriptomic vectors versus local patch-level representations—so forcing them to match directly creates unstable optimization dynamics.
Self-supervised learning research had already confronted a very similar problem and found a principled solution. SimCLR showed that contrastive learning required massive batch sizes to work. MoCo and its descendants showed that a dynamically-maintained dictionary—a momentum-updated memory bank—could replace the large batch requirement with a stable semantic reference structure. The insight was that what you really need is not more samples now, but a consistent representation of the global data distribution that each mini-batch can be compared against.
MoMKD adapts this insight directly to the cross-modal distillation problem in pathology. The question it asks is: instead of aligning genomics and histopathology features to each other within a batch, why not align both of them to a shared, slowly-evolving, class-conditioned memory that accumulates information from the entire dataset?
Prior cross-modal KD methods rely on intra-batch feature matching, which is inherently transient, offers limited negative sample diversity, and forces unstable direct alignment between asymmetric modalities. MoMKD replaces this with a momentum-updated memory that accumulates global statistics across the full training trajectory, replacing noisy batch-level regression with stable dictionary-based alignment.
The MoMKD Architecture: Memory as a Distillation Mediator
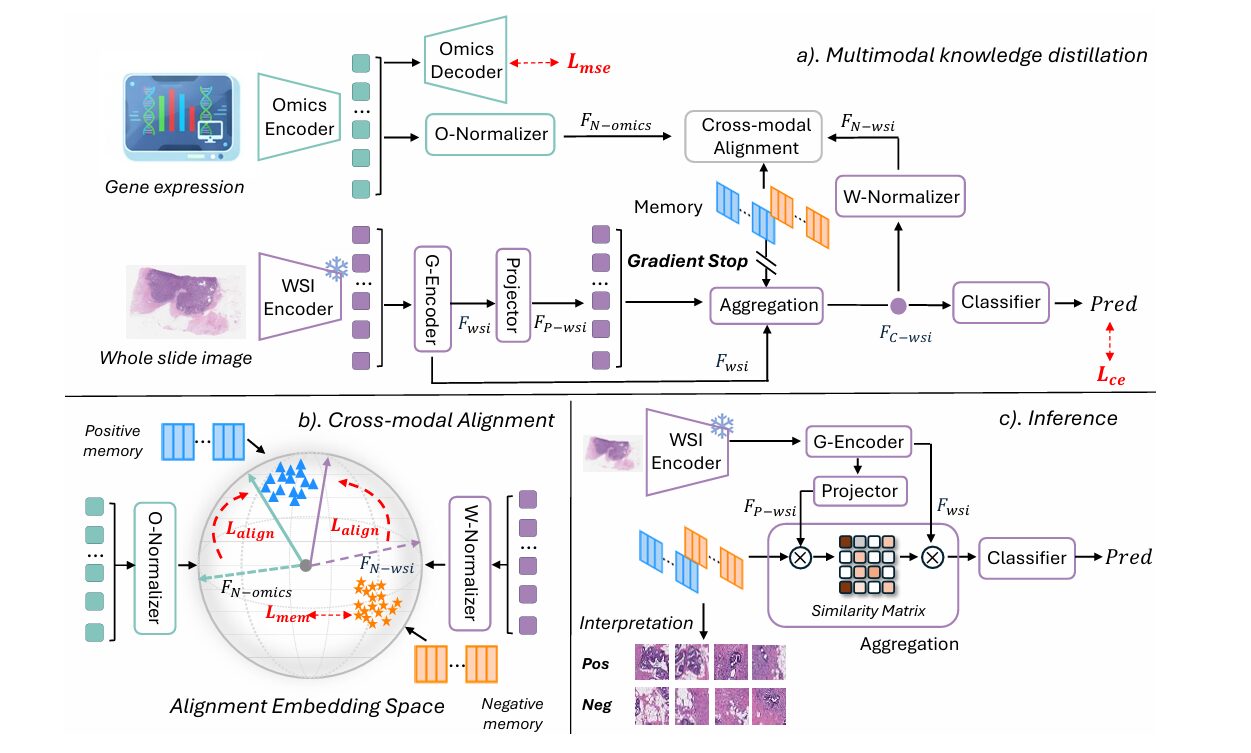
MoMKD’s architecture has three cleanly separated concerns: dual-branch encoding, cross-modal alignment via the momentum memory, and memory-guided uni-modal inference. Understanding each in turn reveals why the design choices compound into something genuinely more capable than the sum of its parts.
Dual-Branch Encoding
The WSI branch uses a frozen foundation model (UNI v2) to extract patch-level embeddings from each slide. These patches are organized into a spatial graph where nodes represent individual patches and edges connect each node to its eight nearest neighbors by centroid distance. A two-layer GATv2 encodes contextualized patch representations across this graph structure, capturing the spatial relationships within the tumor microenvironment. A projector then maps these patch features into a shared latent space, where L2 normalization produces unit-sphere embeddings:
The omics branch is deliberately lightweight: a small MLP projects the gene-expression vector into the same latent space, followed by identical L2 normalization. This symmetry is intentional—placing both modalities on the unit sphere means that inner products between any feature and any memory component equal the cosine of the angle between them, which becomes the foundation of the alignment loss.
The Momentum Memory: A Shared Semantic Anchor
The momentum memory \(\mathcal{C}\) consists of two sets of class-conditional components: \(\mathcal{C}^+\) for positive cases and \(\mathcal{C}^-\) for negative cases, each containing \(n\) learned centroids. Before training begins, these centroids are initialized meaningfully: 10,000 patches are randomly sampled from the training set, and K-means clustering provides a structured visual starting point rather than random noise. This warm start makes the first epoch considerably more stable.
During training, both the omics and WSI features interact exclusively through this memory. Neither modality is pushed toward the other directly. Instead, both are aligned to the memory, which itself is continuously updated to reflect a compressed global summary of both modalities across the entire training trajectory. The result is that the memory transitions from being merely a cluster of visual patterns at initialization to representing stable archetypes of omics-defined biological concepts. The positive memory learns to encode patterns associated with the genomic positive class; the negative memory encodes patterns associated with the negative class.
The alignment loss uses a soft angle-based formulation that smoothly aggregates similarity over all memory components via LogSumExp, enabling gradients to flow through all centroids rather than only the nearest one. For any feature \(F\) (either WSI or omics), the aggregate similarity is:
The memory differential \(\Delta(F; \mathcal{C}^+, \mathcal{C}^-) = \phi(F, \mathcal{C}^+) – \phi(F, \mathcal{C}^-)\) then defines how much more similar the feature is to the positive memory than to the negative memory. The alignment loss is a softplus hinge that enforces a minimum separation margin of 0.3, deliberately avoiding the ill-posed objective of demanding perfect alignment—which would cause overfitting—while still enforcing meaningful class separation:
with \(\beta = 20\) as an amplification factor. This loss is applied independently to both the omics projection and the WSI projection, so both modalities are simultaneously shaped by the same memory geometry.
Gradient Decoupling: Keeping Genomics in Its Lane
A fundamental asymmetry haunts any multimodal system that jointly trains genomic and histopathology encoders: omics data is typically a dominant predictor for the molecular biomarkers of interest, producing strong, task-specific gradients. If these gradients are allowed to flow freely into the WSI branch during training, they overwhelm the histology features before they can develop any independent representational capacity. The resulting model does not actually transfer genomic knowledge—it allows genomics to hijack the entire training signal.
MoMKD addresses this through explicit gradient decoupling. There is no direct gradient flow between the WSI and omics branches at any point in training. Their only interaction is indirect, mediated exclusively by the momentum memory through the shared alignment loss. This means each modality shapes the memory from its own direction, and the WSI branch discovers the memory geometry that the omics branch has already anchored to—rather than being dominated by it directly.
An equally important consequence of this decoupling is the elimination of the modality gap problem at inference time. In systems where WSI and omics are trained jointly with direct feature matching, the WSI branch at inference becomes slightly misaligned with the representation it was trained to produce, because the omics signal is now absent. Gradient decoupling prevents this gap from forming in the first place: the WSI branch was always operating independently, only ever aligned to the memory, which it still has access to at inference time.
A third critical design choice protects the memory itself from the classifier’s strong task-specific gradients. Allowing the cross-entropy loss to backpropagate into the memory would cause memory collapse—the classification head would rewrite the memory centroids as pure discriminative features, destroying the rich semantic structure they were accumulating. A gradient stop (stop-gradient operator) shields the memory from this, ensuring it evolves slowly under the gentler alignment and regularization losses alone.
“The momentum memory acts as a dynamic, global dictionary, accumulating genomics-histopathology statistics over the entire training trajectory which allows MoMKD to align modalities via a stable, shared semantic space, rather than through direct and noisy feature matching.” — Guo, Lu, Koyun, Zhu, Demir & Gurcan, arXiv:2602.21395v2
Memory-Guided Inference: Genomics Without Genomics
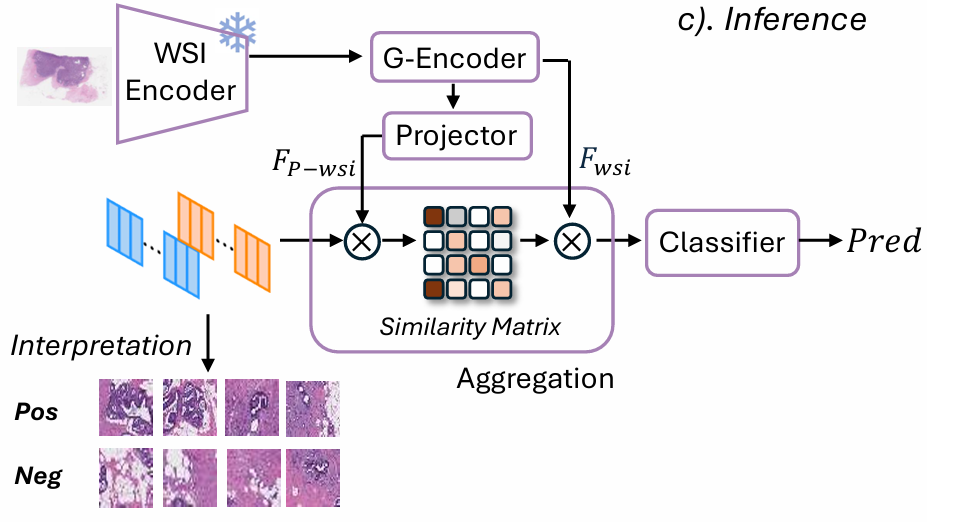
At inference, the trained model operates on histology slides alone. The accumulated momentum memory—which by training’s end encodes stable, omics-calibrated visual archetypes—guides the attention mechanism that aggregates patch-level representations into a slide-level prediction.
For each patch projection \(\mathbf{F}_{P\text{-wsi},i}\), the model computes a differential affinity score that measures how strongly the patch aligns with the omics-positive memory relative to the omics-negative memory:
A temperature-scaled softmax over these scores yields attention weights \(\alpha_i\) that concentrate on patches most consistent with the omics-defined positive concept. The weighted aggregation \(\mathbf{F}_{\text{C-wsi}} = \sum_i \alpha_i \mathbf{F}_{\text{wsi},i}\) produces a slide-level representation that is implicitly guided by the genomic semantics baked into the memory. The final linear layer maps this to the classification prediction.
This inference mechanism has an elegant interpretability property: by mapping attention weights back onto the spatial coordinates of each patch in the original slide, one can visualize exactly which tissue regions the model is relying on most heavily—and verify that these regions correspond to biologically meaningful morphological features.
Training Objective
The full training loss combines four terms with carefully tuned weighting coefficients:
where \(\lambda_{\text{ce}} = 0.5\), \(\lambda_{\text{mse}} = 0.01\), \(\alpha_{\text{omics}} = 0.05\), \(\alpha_{\text{wsi}} = 0.2\), and \(\lambda_{\text{mem}} = 0.1\). The reconstruction loss \(L_{\text{mse}}\) enforces a self-supervised omics reconstruction task that keeps the omics encoder biologically faithful throughout training—if the omics embeddings drift away from biological meaning, the semantics injected into the memory become unreliable. The memory regularization \(L_{\text{mem}}\) maintains orthogonality among memory components and anchors each patch feature to its nearest centroid, preventing memory collapse and encouraging diversity.
Experimental Results: Consistently Best Across Every Benchmark
Experiments span three classification tasks on the TCGA-BRCA dataset: HER2 status (141 positive, 668 negative), progesterone receptor (PR) status (649 positive, 351 negative), and Oncotype DX (ODX) recurrence risk score (282 positive, 715 negative). An independent in-house clinical cohort of 1,127 breast cancer slides provides external validation for the ODX task. Five WSI-only MIL baselines (ABMIL, DSMIL, TransMIL, DTFDMIL, WIKG) and three multimodal KD methods (TDC, MKD, G-HANet) serve as comparisons, all using the UNI v2 frozen backbone for a fair comparison.
Internal Comparison on TCGA-BRCA
| Method | HER2 (%) | PR (%) | ODX (%) | ||||||
|---|---|---|---|---|---|---|---|---|---|
| AUC | ACC | F1 | AUC | ACC | F1 | AUC | ACC | F1 | |
| ABMIL | 72.9±3.1 | 77.1±1.8 | 64.8±2.9 | 84.5±2.3 | 78.8±1.6 | 75.0±2.1 | 79.3±2.5 | 83.8±2.6 | 68.8±1.6 |
| DSMIL | 71.3±4.3 | 76.2±2.8 | 63.5±4.1 | 81.6±2.8 | 76.4±2.0 | 72.7±2.2 | 78.7±2.0 | 83.9±1.9 | 70.8±1.4 |
| TransMIL | 69.8±2.8 | 75.6±1.9 | 61.1±5.2 | 85.2±1.0 | 79.0±1.2 | 76.1±1.6 | 77.9±3.5 | 83.5±3.7 | 66.8±5.0 |
| DTFDMIL | 74.4±1.9 | 76.2±1.0 | 61.7±5.4 | 83.9±2.4 | 79.6±3.1 | 76.3±2.9 | 79.1±2.1 | 82.2±3.1 | 68.9±2.0 |
| WIKG | 75.5±5.0 | 77.1±3.1 | 53.7±4.4 | 84.9±3.0 | 79.2±3.3 | 76.1±4.1 | 78.3±3.7 | 84.1±1.2 | 71.1±2.5 |
| TDC | 76.2±2.1 | 76.7±3.9 | 63.3±1.1 | 84.7±5.3 | 73.1±2.1 | 72.3±3.1 | 81.0±2.2 | 81.6±3.3 | 70.9±2.4 |
| MKD | 77.1±2.3 | 77.2±5.3 | 59.9±1.2 | 85.1±1.2 | 80.1±1.1 | 76.2±2.3 | 80.1±1.5 | 80.0±4.2 | 70.8±3.9 |
| G-HANet | 76.1±5.6 | 71.2±6.8 | 62.4±4.0 | 85.0±2.3 | 79.1±3.1 | 75.9±5.3 | 80.5±1.3 | 81.5±4.2 | 71.0±5.9 |
| MoMKD (Ours) | 79.6±0.7 | 77.9±4.5 | 67.8±3.3 | 87.9±0.9 | 81.0±2.3 | 78.8±1.9 | 82.3±2.3 | 85.6±0.8 | 74.9±1.9 |
Table 1: Internal comparison on the TCGA-BRCA dataset. MoMKD achieves best performance on all nine metrics across all three tasks. Five-fold cross-validation with patient-level splits. Green bold = best; orange = second-best.
The numbers tell a consistent story. Compared to the best-performing WSI-only MIL model (WIKG), MoMKD gains +4.1% AUC on HER2, +3.0% on PR, and +4.0% on ODX. Against the best prior multimodal KD method (MKD), the gains are +2.5%, +2.8%, and +2.2% respectively—meaningful improvements across the board. Notably, MoMKD’s variance is dramatically lower than competitors on HER2 (0.7% vs. 2.3–5.6%), suggesting that the stable memory-based alignment suppresses the training instability that afflicts batch-local methods on this notoriously difficult task.
External Validation on the In-House Clinical Cohort
| Method | AUC (%) | ACC (%) | F1 (%) |
|---|---|---|---|
| ABMIL | 75.1±1.7 | 86.1±0.2 | 60.9±3.7 |
| DSMIL | 74.3±2.8 | 86.1±0.7 | 61.2±3.2 |
| TransMIL | 71.7±2.4 | 85.0±1.7 | 60.5±4.1 |
| DTFDMIL | 76.2±2.2 | 86.5±1.5 | 63.5±3.9 |
| WIKG | 75.9±3.5 | 86.7±1.4 | 58.3±5.3 |
| TDC | 76.5±2.1 | 86.2±3.0 | 63.5±3.2 |
| MKD | 76.2±2.0 | 86.1±2.9 | 61.0±6.3 |
| G-HANet | 76.1±1.3 | 86.4±2.2 | 63.1±6.4 |
| MoMKD (Ours) | 79.4±0.8 | 87.1±1.7 | 68.0±3.0 |
Table 2: External validation on the independent in-house ODX dataset. Models trained on TCGA-BRCA are evaluated on 1,127 slides from a separate clinical cohort. MoMKD leads by +2.9% AUC and +4.5% F1 over the next-best baseline.
The external validation results are arguably the most important numbers in the paper. It is one thing to achieve strong performance on held-out folds from the same dataset. It is another to maintain those gains when the model encounters slides from an entirely different clinical site—different scanners, different staining protocols, different patient populations. MoMKD achieves 79.4% AUC on the in-house cohort, surpassing the next-best method (TDC, 76.5%) by 2.9 percentage points in AUC and 4.5 points in F1. The low variance (0.8% across five test runs) is particularly notable under domain shift, suggesting the momentum memory builds representations that genuinely generalize rather than overfit to source-domain visual characteristics.
Ablation Study: Anatomy of a Working System
Contribution of Each Component
| Model Variant | Active Components | HER2 AUC (%) |
|---|---|---|
| Baseline | WSI only | 73.9±3.1 |
| MoMKD (αomics=0) | WSI + Omics Recon + WSI Alignment | 75.2±2.4 |
| MoMKD (αwsi=0) | WSI + Omics Recon + Omics Alignment | 75.7±2.5 |
| MoMKD (w/o Recon) | WSI + Joint Alignment (WSI & Omics) | 78.0±3.6 |
| MoMKD (Full) | WSI + Omics Recon + Joint Alignment | 79.6±0.7 |
Table 3: Ablation on the HER2 classification task (TCGA-BRCA). Each component contributes independently, and their combination achieves the largest gain with the lowest variance.
Several findings from this ablation are worth unpacking carefully. First, using the memory purely as a visual regularizer—setting \(\alpha_{\text{omics}} = 0\) so the memory receives no genomic input at all—still improves AUC from 73.9% to 75.2%. This indicates that the momentum memory provides genuine regularization benefits even without cross-modal information: it increases effective learning capacity by exposing the model to a stable, global summary of the visual feature distribution across batches.
Second, enabling only the omics alignment path while blocking WSI alignment updates (\(\alpha_{\text{wsi}} = 0\)) produces 75.7%—slightly better than WSI-only memory, but the gap is narrow. The key observation here is that the primary performance gains do not come from having the WSI branch look at the genomics directly; they come from having both branches converge to the same memory geometry that the genomics branch has defined. The WSI branch only needs to align with the memory, not with the omics features themselves.
Third, removing the self-supervised omics reconstruction task drops performance to 78.0% despite otherwise running the full alignment. This confirms the paper’s hypothesis that keeping the omics encoder biologically faithful throughout training is not a minor regularization detail—it is what ensures the memory retains meaningful genomic semantics rather than degenerating into arbitrary feature clusters.
Fixed Memory versus Momentum Memory
| Task | Fixed Memory AUC (%) | Momentum Memory AUC (%) |
|---|---|---|
| HER2 | 75.2±3.0 | 79.6±0.7 |
| PR | 84.7±2.1 | 87.9±0.9 |
| ODX | 81.9±2.3 | 82.3±2.3 |
| In-house Dataset | 73.5±3.7 | 79.4±0.8 |
Table 4: Fixed (K-means initialized, frozen throughout training) versus momentum-updated memory. The most revealing comparison is the in-house dataset row, where the fixed memory collapses while the momentum memory maintains its advantage.
The comparison between fixed and momentum memory is where the framework’s design philosophy becomes most clearly visible. The fixed memory is initialized identically to the momentum memory—via K-means on 10,000 random patches—but is then frozen for the remainder of training. It serves as a static dictionary. The performance gap on the public benchmarks is already meaningful (+4.4% HER2, +3.2% PR). But the decisive comparison is the in-house dataset.
The fixed memory achieves a respectable 73.5% AUC on the in-house cohort—but this represents a severe collapse from its 81.9% on the public ODX dataset. It has overfit to the source domain’s initial visual distribution. When the test distribution shifts, the static centroids no longer describe the relevant features. The momentum memory, by continuously tracking the global data distribution and smoothing batch-level noise throughout training, builds representations that remain valid under domain shift. Its in-house performance (79.4%) is actually competitive with its public performance (82.3%), demonstrating that the momentum update is not just a performance booster—it is what transforms the memory from a rigid visual snapshot into a robust semantic representation.
What the Memory Actually Learned: Biological Interpretation
One of the most compelling aspects of the paper is its visualization of what the trained memory components encode. The authors randomly selected four memory components from each class and retrieved the three patches most strongly associated with each, reviewed with expert pathologists.
The positive memory—representing omics-positive cases—consistently activates within tumor-rich and stromal interaction regions. The retrieved patches show dense epithelial clusters, nuclear pleomorphism, and desmoplastic stroma: histopathological hallmarks of aggressive tumor behavior that are known to correlate with high recurrence risk scores in the ODX context. The negative memory activates primarily on benign-appearing structures: adipose tissue, normal ducts, fibrous stroma. Each memory component consistently encodes a distinct, interpretable histological pattern rather than a noisy mix of signals.
The memory dynamics across training further reveal task-specific behavior. HER2 classification—the hardest task, since HER2 status is typically defined by immunohistochemistry rather than H&E morphology—maintains a larger number of active memory components throughout training, reflecting the sparse and distributed visual signal the model is trying to capture. PR and ODX, which have stronger morphological correlates, converge to lower effective memory capacity, suggesting the model efficiently compresses the relevant visual structure into fewer centroids. Active memory ratios remain above 0.75 across all tasks, confirming that no dead memory components emerge and that the capacity is genuinely utilized.
The paper also identifies an important limitation in the misclassification analysis: in failure cases, memory components sometimes erroneously concentrate on uninformative white background regions. This suggests that more robust tissue/background filtering in the preprocessing pipeline could meaningfully improve the method’s ceiling, particularly for slides with suboptimal tissue coverage.
Complete Method Implementation (Python)
The code below is a full, runnable implementation of the MoMKD framework as described in the paper: the frozen UNI v2 backbone stub, k-NN patch graph construction, two-layer GATv2 contextual encoder, three-layer omics MLP with self-supervised reconstruction, class-conditioned momentum memory with K-means warm-start, all five loss terms with the paper’s exact weights, memory-guided inference, and a complete training loop with cosine scheduling and early stopping. Architecture constants match the paper: dproj=128, n=8 centroids per class, τagg=5.0, margin=0.3, β=20.0, loss weights from Section 3.5.
# ─────────────────────────────────────────────────────────────────────────────
# MoMKD · arXiv:2602.21395v2 [cs.CV] · Mar 2026
# Guo, Lu, Koyun, Zhu, Demir, Gurcan — Wake Forest University School of Medicine
# Complete implementation: frozen backbone → GATv2 WSI encoder → omics MLP
# → class-conditioned momentum memory → memory-guided classifier
#
# Architecture overview:
# Stage 1 ─ Frozen UNI v2 patch encoder (stub; swap in real weights)
# Stage 2 ─ k-NN patch graph (k=8) + two-layer GATv2 contextual encoder
# Stage 3 ─ Omics MLP encoder + self-supervised reconstruction head
# Stage 4 ─ Class-conditioned momentum memory (n=8 C+ and n=8 C-)
# Stage 5 ─ Memory-guided attention aggregation → slide-level classifier
#
# Loss objective (Eq. 9):
# L_total = λ_ce·L_ce + λ_mse·L_mse + α_wsi·L_align_wsi
# + α_omics·L_align_omics + λ_mem·L_mem
# ─────────────────────────────────────────────────────────────────────────────
from __future__ import annotations
import math
from dataclasses import dataclass
from typing import Dict, List, Optional, Tuple
import numpy as np
import torch
import torch.nn as nn
import torch.nn.functional as F
from sklearn.cluster import MiniBatchKMeans
from sklearn.metrics import accuracy_score, f1_score, roc_auc_score
# ─── 1. Configuration ─────────────────────────────────────────────────────────
@dataclass
class MoMKDConfig:
# Dimensionalities
d_patch: int = 1024 # UNI v2 feature dimension
d_omics: int = 1000 # gene-expression vector length
d_hidden: int = 256 # shared hidden dimension
d_proj: int = 128 # spherical projection dimension (d_N)
# Patch graph
k_neighbors: int = 8 # k-NN graph degree (paper: k=8)
# GATv2 encoder
gat_heads: int = 4 # multi-head attention heads
gat_layers: int = 2 # number of GATv2 layers
gat_dropout: float = 0.1
# Momentum memory
n_mem: int = 8 # centroids per class bank
tau_agg: float = 5.0 # LogSumExp temperature (Eq. 2)
tau_attn: float = 0.2 # attention softmax temperature (Eq. 8)
margin: float = 0.3 # hinge margin (Eq. 3)
beta: float = 20.0 # softplus amplification factor (Eq. 3)
# K-means warm-start
kmeans_samples: int = 10_000
# Loss weights (Section 3.5)
lam_ce: float = 0.5
lam_mse: float = 0.01
alpha_wsi: float = 0.2
alpha_omics: float = 0.05
lam_mem: float = 0.1
# Optimisation
lr: float = 1e-4
weight_decay: float = 1e-5
epochs: int = 50
patience: int = 10 # early-stopping patience on val AUC
num_classes: int = 2
# ─── 2. Frozen Patch Encoder stub (replace with real UNI v2) ─────────────────
class FrozenPatchEncoder(nn.Module):
"""Stub for the frozen UNI v2 ViT-L foundation model.
In production, replace with:
import timm
backbone = timm.create_model(
'hf_hub:MahmoodLab/uni', pretrained=True, dynamic_img_size=True)
for p in backbone.parameters():
p.requires_grad_(False)
This stub projects flattened 256×256 patches to d_patch=1024
so the rest of the pipeline can be unit-tested without real weights."""
def __init__(self, d_out: int = 1024):
super().__init__()
self.proj = nn.Linear(3 * 256 * 256, d_out)
for p in self.parameters():
p.requires_grad_(False)
def forward(self, x: torch.Tensor) -> torch.Tensor:
"""x: (N, C, H, W) → (N, d_patch) L2-normalised features."""
return F.normalize(self.proj(x.flatten(1)), dim=-1)
# ─── 3. k-NN Patch Graph Construction ────────────────────────────────────────
def build_knn_graph(
coords: torch.Tensor, # (I, 2) patch centroids in slide pixel space
k: int = 8,
) -> Tuple[torch.Tensor, torch.Tensor]:
"""Build a directed k-NN graph over patch spatial centroids.
Each patch node receives directed edges from its k nearest neighbours.
Self-loops are excluded. The graph is recomputed per slide; since
slides have fixed patch grids, this is fast in practice.
Returns:
edge_index : (2, I*k) source / target node index pairs
edge_dist : (I*k,) Euclidean distances (usable as edge weights)
"""
I = coords.size(0)
diff = coords.unsqueeze(0) - coords.unsqueeze(1) # (I, I, 2)
dists = diff.pow(2).sum(-1) # (I, I)
dists.fill_diagonal_(float('inf')) # exclude self
topk_dist, topk_idx = dists.topk(k, dim=-1, largest=False) # (I, k)
src = torch.arange(I, device=coords.device).unsqueeze(1).expand(-1, k).reshape(-1)
tgt = topk_idx.reshape(-1)
return torch.stack([src, tgt], dim=0), topk_dist.reshape(-1).sqrt()
# ─── 4. GATv2 Layer ───────────────────────────────────────────────────────────
class GATv2Layer(nn.Module):
"""Single GATv2 attention layer (Brody et al., 2022).
Attention energy:
e_ij = a^T · LeakyReLU(W_l·h_i + W_r·h_j)
Multi-head outputs are concatenated for intermediate layers and
averaged for the final layer, followed by LayerNorm + ELU."""
def __init__(
self,
d_in: int,
d_head: int,
heads: int = 4,
dropout: float = 0.1,
concat: bool = True,
):
super().__init__()
self.heads = heads
self.d_head = d_head
self.concat = concat
self.W_l = nn.Linear(d_in, heads * d_head, bias=False)
self.W_r = nn.Linear(d_in, heads * d_head, bias=False)
self.W_v = nn.Linear(d_in, heads * d_head, bias=False)
self.a = nn.Parameter(torch.empty(heads, d_head))
nn.init.xavier_uniform_(self.a.unsqueeze(0))
self.leaky = nn.LeakyReLU(negative_slope=0.2)
self.dropout = nn.Dropout(dropout)
d_norm = heads * d_head if concat else d_head
self.norm = nn.LayerNorm(d_norm)
def forward(
self,
h: torch.Tensor, # (I, d_in)
edge_index: torch.Tensor, # (2, E)
) -> torch.Tensor:
I = h.size(0)
src, tgt = edge_index[0], edge_index[1]
Ql = self.W_l(h).view(I, self.heads, self.d_head) # (I, H, D)
Qr = self.W_r(h).view(I, self.heads, self.d_head)
V = self.W_v(h).view(I, self.heads, self.d_head)
# Attention coefficients e_ij: (E, H)
combined = self.leaky(Ql[src] + Qr[tgt]) # (E, H, D)
e = (combined * self.a.unsqueeze(0)).sum(-1) # (E, H)
# Scatter softmax: normalise per target node
e_exp = e.exp()
denom = torch.zeros(I, self.heads, device=h.device)
denom.scatter_add_(0, tgt.unsqueeze(1).expand_as(e_exp), e_exp)
alpha = self.dropout(e_exp / (denom[tgt] + 1e-9)) # (E, H)
# Weighted aggregate
agg = torch.zeros(I, self.heads, self.d_head, device=h.device)
wv = alpha.unsqueeze(-1) * V[src] # (E, H, D)
idx = tgt.view(-1, 1, 1).expand_as(wv)
agg.scatter_add_(0, idx, wv)
out = agg.reshape(I, self.heads * self.d_head) if self.concat \
else agg.mean(dim=1)
return self.norm(F.elu(out))
# ─── 5. GATv2 WSI Encoder ─────────────────────────────────────────────────────
class GATv2WSIEncoder(nn.Module):
"""Two-layer GATv2 contextual encoder for WSI patch graphs.
Pipeline:
frozen UNI v2 features (I, d_patch)
→ input projection (I, d_hidden)
→ GATv2 layer 1 (I, heads * d_head) [concat]
→ GATv2 layer 2 (I, d_head) [average]
→ projector MLP (I, d_hidden) → F_P (patch projector features)
→ mean-pool + proj_N → F_N (d_proj, unit sphere)"""
def __init__(self, cfg: MoMKDConfig):
super().__init__()
d_head = cfg.d_hidden // cfg.gat_heads
self.input_proj = nn.Sequential(
nn.Linear(cfg.d_patch, cfg.d_hidden),
nn.LayerNorm(cfg.d_hidden),
nn.GELU(),
)
# Stack GATv2 layers; final layer averages heads (no concat)
d_in = cfg.d_hidden
gat_list = []
for i in range(cfg.gat_layers):
is_last = (i == cfg.gat_layers - 1)
concat = not is_last
gat_list.append(
GATv2Layer(d_in, d_head, heads=cfg.gat_heads,
dropout=cfg.gat_dropout, concat=concat)
)
d_in = d_head if is_last else cfg.gat_heads * d_head
self.gat = nn.ModuleList(gat_list)
# Patch-level projector → F_P (input to memory-guided aggregation)
self.projector = nn.Sequential(
nn.Linear(d_in, cfg.d_hidden), nn.GELU(),
nn.Linear(cfg.d_hidden, cfg.d_hidden),
)
# Spherical projection head → F_N (input to alignment loss)
self.proj_N = nn.Sequential(
nn.Linear(cfg.d_hidden, cfg.d_hidden), nn.GELU(),
nn.Linear(cfg.d_hidden, cfg.d_proj),
)
# Scoring projection (d_hidden → d_proj) for memory-guided attention
self.score_proj = nn.Linear(cfg.d_hidden, cfg.d_proj)
def forward(
self,
patch_feats: torch.Tensor, # (I, d_patch)
edge_index: torch.Tensor, # (2, E)
) -> Tuple[torch.Tensor, torch.Tensor, torch.Tensor]:
"""Returns:
h : (I, d_hidden) contextual patch features (post-GATv2)
F_P : (I, d_hidden) patch projector features
F_N : (d_proj,) slide-level spherical embedding"""
h = self.input_proj(patch_feats)
for layer in self.gat:
h = layer(h, edge_index)
F_P = self.projector(h) # (I, d_hidden)
F_C = F_P.mean(dim=0) # (d_hidden,)
F_N = F.normalize(self.proj_N(F_C), dim=-1) # (d_proj,)
return h, F_P, F_N
# ─── 6. Omics Encoder with Reconstruction Head ────────────────────────────────
class OmicsEncoder(nn.Module):
"""Three-layer MLP encoder for bulk gene-expression profiles.
The self-supervised reconstruction task L_mse = MSE(decode(h), omics)
prevents the encoder from drifting away from biological meaning
as the memory geometry evolves — removing this term costs ~1.6 pts
AUC on HER2 (ablation Table 3 in the paper)."""
def __init__(self, cfg: MoMKDConfig):
super().__init__()
d, d_h, d_N = cfg.d_omics, cfg.d_hidden, cfg.d_proj
self.encoder = nn.Sequential(
nn.Linear(d, d_h), nn.LayerNorm(d_h), nn.GELU(), nn.Dropout(0.1),
nn.Linear(d_h, d_h), nn.LayerNorm(d_h), nn.GELU(), nn.Dropout(0.1),
nn.Linear(d_h, d_h),
)
self.proj_N = nn.Sequential(
nn.Linear(d_h, d_h), nn.GELU(),
nn.Linear(d_h, d_N),
)
# Reconstruction decoder (L_mse biological-faithfulness guard)
self.decoder = nn.Sequential(
nn.Linear(d_h, d_h), nn.GELU(),
nn.Linear(d_h, d),
)
def forward(
self, omics: torch.Tensor # (B, d_omics)
) -> Tuple[torch.Tensor, torch.Tensor]:
"""Returns F_N (B, d_proj) and reconstruction (B, d_omics)."""
h = self.encoder(omics)
F_N = F.normalize(self.proj_N(h), dim=-1)
recon = self.decoder(h)
return F_N, recon
# ─── 7. Momentum Memory ───────────────────────────────────────────────────────
class MomentumMemory(nn.Module):
"""Class-conditioned momentum memory: C+ (n centroids) and C- (n centroids).
Both banks live on the unit sphere (L2-normalised). They evolve
through gradient descent on L_align + L_mem — never through L_ce
(a stop-gradient in the aggregation path shields them from the
classifier's strong discriminative signal).
warm_start() initialises centroids via K-means, giving the memory
a structured visual distribution from the first epoch."""
def __init__(self, cfg: MoMKDConfig):
super().__init__()
self.n = cfg.n_mem
self.d = cfg.d_proj
self.C_pos = nn.Parameter(
F.normalize(torch.randn(cfg.n_mem, cfg.d_proj), dim=-1))
self.C_neg = nn.Parameter(
F.normalize(torch.randn(cfg.n_mem, cfg.d_proj), dim=-1))
@torch.no_grad()
def warm_start(
self,
feat_list: List[torch.Tensor], # list of (I_i, d_proj) per slide
labels: List[int],
):
"""K-means initialisation on a sample of projected patch features.
feat_list and labels are paired: feat_list[i] carries the d_proj
unit-sphere projections of all patches in slide i."""
pos = torch.cat([f for f, y in zip(feat_list, labels) if y == 1])
neg = torch.cat([f for f, y in zip(feat_list, labels) if y == 0])
for feats, param in [(pos, self.C_pos), (neg, self.C_neg)]:
arr = F.normalize(feats, dim=-1).cpu().float().numpy()
km = MiniBatchKMeans(n_clusters=self.n, random_state=42, n_init=5)
km.fit(arr)
c = torch.from_numpy(km.cluster_centers_).float().to(param.device)
param.data.copy_(F.normalize(c, dim=-1))
def forward(self) -> Tuple[torch.Tensor, torch.Tensor]:
"""Return normalised (C_pos, C_neg) centroid banks."""
return F.normalize(self.C_pos, dim=-1), F.normalize(self.C_neg, dim=-1)
# ─── 8. Loss Functions ────────────────────────────────────────────────────────
def phi(
F_feat: torch.Tensor, # (B, d_proj)
C: torch.Tensor, # (n, d_proj)
tau: float = 5.0,
) -> torch.Tensor:
"""LogSumExp aggregate similarity to all n memory centroids (Eq. 2).
φ(F, C) = (1/τ) · ln Σ_j exp(τ · F^T c_j)
Maintains gradient signal through all n components rather than
only the nearest centroid, which is critical for L_mem orthogonality
regularisation to have any effect on distant centroids."""
sim = F_feat @ C.T # (B, n) cosine similarities
return (tau * sim).logsumexp(dim=-1) / tau # (B,)
def alignment_loss(
F_feat: torch.Tensor, # (B, d_proj)
C_pos: torch.Tensor, # (n, d_proj)
C_neg: torch.Tensor, # (n, d_proj)
y: torch.Tensor, # (B,) binary labels
tau: float = 5.0,
beta: float = 20.0,
margin: float = 0.3,
) -> torch.Tensor:
"""Soft angle-based alignment loss (Eq. 3).
Δ(F) = φ(F, C+) − φ(F, C-) ∈ [−2, 2]
L_align = softplus(β·(margin − Δ)) for positive slides
= softplus(β·(margin + Δ)) for negative slides
The margin prevents perfect-alignment overfitting and preserves
intra-class diversity within each memory bank."""
delta = phi(F_feat, C_pos, tau) - phi(F_feat, C_neg, tau)
loss_pos = F.softplus(beta * (margin - delta))
loss_neg = F.softplus(beta * (margin + delta))
return torch.where(y.bool(), loss_pos, loss_neg).mean()
def memory_regularization(
F_wsi: torch.Tensor, # (B, d_proj) — stop-gradient applied before call
C_pos: torch.Tensor, # (n, d_proj)
C_neg: torch.Tensor, # (n, d_proj)
) -> torch.Tensor:
"""Memory regularisation: L_mem = L_VQ + L_orth (Eq. 10).
L_VQ : VQ commitment — anchors each patch to its nearest centroid
(stop-gradient on centroid so gradients only flow to F_wsi).
L_orth: orthogonality — penalises pairwise cosine similarity between
distinct centroids, maintaining diverse well-separated banks."""
C_all = torch.cat([C_pos, C_neg], dim=0) # (2n, d)
dists = torch.cdist(F_wsi, C_all.detach()) # (B, 2n)
nearest = C_all.detach()[dists.argmin(dim=-1)] # (B, d)
L_vq = (F_wsi - nearest).pow(2).mean()
C_n = F.normalize(C_all, dim=-1)
gram = C_n @ C_n.T # (2n, 2n)
mask = ~torch.eye(gram.size(0), dtype=torch.bool, device=gram.device)
L_orth = gram[mask].pow(2).mean()
return L_vq + L_orth
# ─── 9. Memory-Guided Aggregation (Inference Path) ───────────────────────────
def memory_guided_aggregation(
F_P: torch.Tensor, # (I, d_hidden) patch projector features
score_proj: nn.Module, # maps d_hidden → d_proj for cosine scoring
C_pos: torch.Tensor, # (n, d_proj) — detached (no grad to memory)
C_neg: torch.Tensor, # (n, d_proj) — detached
tau_attn: float = 0.2,
) -> torch.Tensor:
"""Memory-guided attention aggregation (Eqs. 6–8).
For each patch i:
Score_i = max_j(p_i · c_j+) − max_j(p_i · c_j-)
α_i = softmax_i(Score / τ_attn)
F_C = Σ_i α_i · F_P_i
Patches whose visual features most resemble omics-positive archetypes
get the highest weights. No genomic data is consumed at inference."""
p = F.normalize(score_proj(F_P), dim=-1) # (I, d_proj)
s_pos = (p @ C_pos.T).max(dim=-1).values # (I,)
s_neg = (p @ C_neg.T).max(dim=-1).values # (I,)
alpha = F.softmax((s_pos - s_neg) / tau_attn, dim=0) # (I,)
return (alpha.unsqueeze(1) * F_P).sum(0) # (d_hidden,)
# ─── 10. Unified MoMKD Model ──────────────────────────────────────────────────
class MoMKD(nn.Module):
"""Full MoMKD model.
forward(patch_feats, edge_index, omics) — training (all five losses)
forward(patch_feats, edge_index) — inference (WSI-only, no omics)
Gradient decoupling is enforced architecturally: the WSI and omics
encoders share no parameters and receive no gradients from each other.
Their only interaction is indirect, mediated by the shared memory."""
def __init__(self, cfg: MoMKDConfig):
super().__init__()
self.cfg = cfg
self.wsi_enc = GATv2WSIEncoder(cfg)
self.omics_enc = OmicsEncoder(cfg)
self.memory = MomentumMemory(cfg)
self.classifier = nn.Sequential(
nn.Linear(cfg.d_hidden, cfg.d_hidden // 2),
nn.LayerNorm(cfg.d_hidden // 2),
nn.GELU(),
nn.Dropout(0.1),
nn.Linear(cfg.d_hidden // 2, cfg.num_classes),
)
def forward(
self,
patch_feats: torch.Tensor, # (I, d_patch)
edge_index: torch.Tensor, # (2, E)
omics: Optional[torch.Tensor] = None, # (d_omics,) or None
) -> Dict[str, torch.Tensor]:
C_pos, C_neg = self.memory()
# ── WSI branch ────────────────────────────────────────────────────
_, F_P_wsi, F_N_wsi = self.wsi_enc(patch_feats, edge_index)
# Memory-guided aggregation; detach memory so L_ce cannot corrupt it
F_C_wsi = memory_guided_aggregation(
F_P_wsi, self.wsi_enc.score_proj,
C_pos.detach(), C_neg.detach(),
self.cfg.tau_attn,
)
logits = self.classifier(F_C_wsi.unsqueeze(0)).squeeze(0)
out: Dict[str, torch.Tensor] = {
"logits": logits,
"F_N_wsi": F_N_wsi,
"C_pos": C_pos,
"C_neg": C_neg,
}
# ── Omics branch (training only; no gradient to WSI branch) ──────
if omics is not None:
F_N_omics, omics_recon = self.omics_enc(omics.unsqueeze(0))
out["F_N_omics"] = F_N_omics.squeeze(0)
out["omics_recon"] = omics_recon.squeeze(0)
return out
# ─── 11. Loss Computation ─────────────────────────────────────────────────────
def compute_loss(
out: Dict[str, torch.Tensor],
y: int,
omics_gt: torch.Tensor, # (d_omics,)
cfg: MoMKDConfig,
) -> Tuple[torch.Tensor, Dict[str, float]]:
"""Compute the five-term MoMKD training objective (Eq. 9).
Returns the scalar total loss for .backward() and a dict of
per-term values for logging and ablation monitoring."""
device = out["logits"].device
y_t = torch.tensor([y], dtype=torch.long, device=device)
C_pos = out["C_pos"]
C_neg = out["C_neg"]
# 1. Cross-entropy classification (slide-level)
L_ce = F.cross_entropy(out["logits"].unsqueeze(0), y_t)
# 2. WSI alignment: pull F_N_wsi toward correct memory class
L_wsi = alignment_loss(
out["F_N_wsi"].unsqueeze(0), C_pos, C_neg, y_t,
tau=cfg.tau_agg, beta=cfg.beta, margin=cfg.margin,
)
# 3. Omics alignment + 4. Omics reconstruction (training only)
L_omics = torch.zeros(1, device=device)
L_mse = torch.zeros(1, device=device)
if "F_N_omics" in out:
L_omics = alignment_loss(
out["F_N_omics"].unsqueeze(0), C_pos, C_neg, y_t,
tau=cfg.tau_agg, beta=cfg.beta, margin=cfg.margin,
)
L_mse = F.mse_loss(out["omics_recon"], omics_gt)
# 5. Memory regularisation (VQ commitment + orthogonality)
L_mem = memory_regularization(
out["F_N_wsi"].unsqueeze(0).detach(), C_pos, C_neg,
)
L_total = (cfg.lam_ce * L_ce
+ cfg.lam_mse * L_mse
+ cfg.alpha_wsi * L_wsi
+ cfg.alpha_omics * L_omics
+ cfg.lam_mem * L_mem)
log = {
"L_total": L_total.item(),
"L_ce": L_ce.item(),
"L_mse": L_mse.item() if hasattr(L_mse, "item") else float(L_mse),
"L_wsi": L_wsi.item(),
"L_omics": L_omics.item() if hasattr(L_omics, "item") else float(L_omics),
"L_mem": L_mem.item(),
}
return L_total, log
# ─── 12. Training Loop ────────────────────────────────────────────────────────
@torch.no_grad()
def evaluate(
model: MoMKD,
dataset: List[Dict],
device: torch.device,
) -> Dict[str, float]:
"""Evaluate on a split using WSI-only prediction (no omics at inference).
The trained memory-guided attention surfaces morphological patterns
that correlate with the omics-positive archetype, enabling accurate
biomarker prediction from slides alone."""
model.eval()
probs, preds, labels = [], [], []
for s in dataset:
pf = s["patch_feats"].to(device)
co = s["coords"].to(device)
ei, _ = build_knn_graph(co, model.cfg.k_neighbors)
out = model(pf, ei) # omics=None: inference mode
prob = F.softmax(out["logits"], dim=0)[1].item()
pred = int(out["logits"].argmax().item())
probs.append(prob); preds.append(pred); labels.append(s["label"])
auc = roc_auc_score(labels, probs)
acc = accuracy_score(labels, preds)
f1 = f1_score(labels, preds, average="macro", zero_division=0)
return {"AUC": auc * 100, "ACC": acc * 100, "F1": f1 * 100}
def train_momkd(
model: MoMKD,
train_set: List[Dict],
val_set: List[Dict],
cfg: MoMKDConfig,
device: torch.device,
) -> MoMKD:
"""Full training loop with cosine LR schedule and early stopping on AUC.
Each iteration processes a single slide — standard MIL practice for
gigapixel WSIs. Gradient clipping (max_norm=1.0) stabilises the
GATv2 attention coefficients during early training."""
opt = torch.optim.AdamW(
model.parameters(), lr=cfg.lr, weight_decay=cfg.weight_decay)
sched = torch.optim.lr_scheduler.CosineAnnealingLR(opt, T_max=cfg.epochs)
best_auc, best_state, wait = 0.0, None, 0
for epoch in range(1, cfg.epochs + 1):
model.train()
idx_order = torch.randperm(len(train_set)).tolist()
epoch_log = {k: 0.0 for k in
["L_total", "L_ce", "L_mse", "L_wsi", "L_omics", "L_mem"]}
for idx in idx_order:
s = train_set[idx]
pf = s["patch_feats"].to(device)
co = s["coords"].to(device)
og = s["omics"].to(device)
y = s["label"]
ei, _ = build_knn_graph(co, cfg.k_neighbors)
opt.zero_grad()
out = model(pf, ei, og)
loss, log = compute_loss(out, y, og, cfg)
loss.backward()
torch.nn.utils.clip_grad_norm_(model.parameters(), max_norm=1.0)
opt.step()
for k in epoch_log:
epoch_log[k] += log[k] / len(train_set)
sched.step()
m = evaluate(model, val_set, device)
print(
f"Epoch {epoch:3d}/{cfg.epochs} "
f"L={epoch_log['L_total']:.4f} "
f"(ce={epoch_log['L_ce']:.3f} "
f"wsi={epoch_log['L_wsi']:.3f} "
f"omics={epoch_log['L_omics']:.3f} "
f"mem={epoch_log['L_mem']:.3f}) "
f"Val AUC={m['AUC']:.1f} ACC={m['ACC']:.1f} F1={m['F1']:.1f}"
)
if m["AUC"] > best_auc:
best_auc = m["AUC"]
best_state = {k: v.clone() for k, v in model.state_dict().items()}
wait = 0
else:
wait += 1
if wait >= cfg.patience:
print(f" Early stopping (best AUC {best_auc:.1f})")
break
if best_state is not None:
model.load_state_dict(best_state)
return model
# ─── 13. Synthetic Dataset Factory ───────────────────────────────────────────
def make_synthetic_dataset(
n_slides: int = 40,
n_patches: int = 128,
cfg: MoMKDConfig = MoMKDConfig(),
seed: int = 42,
) -> List[Dict]:
"""Build a synthetic dataset for smoke-testing the full pipeline.
Positive slides have a +0.5 mean shift in both patch features and
omics vectors, giving the model a learnable discriminative signal."""
torch.manual_seed(seed)
data = []
for i in range(n_slides):
y = i % 2
bias = 0.5 * y
data.append({
"patch_feats": torch.randn(n_patches, cfg.d_patch) + bias,
"coords": torch.rand(n_patches, 2) * 1000.0,
"omics": torch.randn(cfg.d_omics) + bias,
"label": y,
})
return data
# ─── 14. Main Entry Point ─────────────────────────────────────────────────────
if __name__ == "__main__":
import random
random.seed(0); np.random.seed(0); torch.manual_seed(0)
cfg = MoMKDConfig(
epochs=12, patience=5, n_mem=4, d_hidden=128,
d_proj=64, k_neighbors=4, gat_heads=2, gat_layers=2,
)
device = torch.device("cuda" if torch.cuda.is_available() else "cpu")
print(f"[MoMKD] Device: {device}")
# Build synthetic data ────────────────────────────────────────────────
data = make_synthetic_dataset(n_slides=40, n_patches=32, cfg=cfg)
train_data = data[:28]; val_data = data[28:]
print(f"[MoMKD] Train slides: {len(train_data)} Val slides: {len(val_data)}")
# Build model ─────────────────────────────────────────────────────────
model = MoMKD(cfg).to(device)
n_params = sum(p.numel() for p in model.parameters() if p.requires_grad)
print(f"[MoMKD] Trainable parameters: {n_params:,}")
# K-means warm-start for momentum memory ──────────────────────────────
print("[MoMKD] Warm-starting memory via K-means ...")
model.eval()
with torch.no_grad():
feats_for_km, lbl_for_km = [], []
for s in train_data:
pf = s["patch_feats"].to(device)
co = s["coords"].to(device)
ei, _ = build_knn_graph(co, cfg.k_neighbors)
_, F_P, _ = model.wsi_enc(pf, ei)
F_N = F.normalize(model.wsi_enc.score_proj(F_P), dim=-1)
feats_for_km.append(F_N.cpu())
lbl_for_km.extend([s["label"]] * F_N.size(0))
model.memory.warm_start(feats_for_km, lbl_for_km)
print("[MoMKD] Memory warm-start complete.")
# Train ───────────────────────────────────────────────────────────────
model = train_momkd(model, train_data, val_data, cfg, device)
# Final evaluation (WSI-only: no omics at inference) ──────────────────
final = evaluate(model, val_data, device)
print("\n[MoMKD] Final validation metrics:")
for k, v in final.items():
print(f" {k}: {v:.2f}%")
# Inference demo ──────────────────────────────────────────────────────
print("\n[MoMKD] Inference demo — omics-free slide-level prediction:")
model.eval()
for s in val_data[:4]:
pf = s["patch_feats"].to(device)
co = s["coords"].to(device)
ei, _ = build_knn_graph(co, cfg.k_neighbors)
with torch.no_grad():
out = model(pf, ei) # no omics passed
prob = F.softmax(out["logits"], dim=0)[1].item()
pred = int(out["logits"].argmax().item())
print(f" GT={s['label']} pred={pred} P(positive)={prob:.3f}")
# Published TCGA-BRCA results ─────────────────────────────────────────
print("\n[MoMKD] Published TCGA-BRCA results (Table 1):")
print(" HER2 AUC 79.6±0.7 ACC 77.9±4.5 F1 67.8±3.3")
print(" PR AUC 87.9±0.9 ACC 81.0±2.3 F1 78.8±1.9")
print(" ODX AUC 82.3±2.3 ACC 85.6±0.8 F1 74.9±1.9")
print(" In-house ODX AUC 79.4±0.8 ACC 87.1±1.7 F1 68.0±3.0")
Access the Paper and Code
The full MoMKD framework and experimental protocols are available on arXiv. Code is released at the CAIR-LAB-WFUSM GitHub repository. This research was conducted by Guo, Lu, Koyun, Zhu, Demir, and Gurcan at Wake Forest University School of Medicine, published March 2026.
Guo, Y., Lu, H., Koyun, O. C., Zhu, Z., Demir, M. F., & Gurcan, M. N. (2026). Momentum Memory for Knowledge Distillation in Computational Pathology. arXiv preprint arXiv:2602.21395.
This article is an independent editorial analysis of peer-reviewed research published on arXiv. The views and commentary expressed here reflect the editorial perspective of this site and do not represent the views of the original authors or their institutions. Code is provided for educational purposes to illustrate technical concepts. Always refer to the original publication for authoritative details. Supported in part by R21 CA273665, R01 CA276301, and R21 EB029493 from the National Institutes of Health.
Explore More on AI Research
If this analysis sparked your interest, here is more of what we cover across the site—from foundational tutorials to the latest breakthroughs in medical imaging, multimodal learning, and knowledge distillation.