Reading Twelve Nutrients from a Flash of Light: The Stacked Regression Pipeline Changing Potato Farm Diagnostics
A team at Dalhousie University built a two-layer machine learning pipeline — MRMR feature selection, Lasso base models, and XGBoost meta-learners — that simultaneously estimates twelve macro- and micronutrient concentrations in potato plants from a single vis-NIR spectral scan, cutting the detection time from days to minutes.
Growing potatoes well is, at its core, a chemistry problem. The plant’s yield, disease resistance, and storability hinge on the concentrations of a dozen elements circulating through its tissue — nitrogen driving leaf area, phosphorus enabling root energy, zinc and iron catalysing enzyme cascades that nothing else can substitute for. The trouble is that finding out what any given field actually contains has always meant waiting: collect the petioles, dry them, ship them to a lab, wait a week, pay per sample, and receive numbers that describe conditions that may have already changed. A team at Dalhousie University and the University of Bristol decided that timeline was no longer acceptable — and built a spectroscopic alternative that delivers the same twelve numbers in minutes.
The Chemistry Problem That Spectroscopy Was Made For
Point a near-infrared lamp at a plant leaf and measure how much light bounces back at each wavelength between 400 and 2500 nm. Every chemical bond in the tissue — C–H, N–H, O–H, the carbon backbones of chlorophyll and cell walls — absorbs and reflects at characteristic frequencies. The resulting spectrum is, in principle, a fingerprint of the leaf’s entire chemical composition, including the macro- and micronutrients it contains. The catch has always been extracting clean, reliable signals for specific elements from a spectrum that reflects everything at once.
Prior work by Reem Abukmeil, Ahmad Al-Mallahi, and Felipe Campelo — published in 2022 — demonstrated that this extraction is possible for individual nutrients using elastic net regression on fresh and dried potato leaves. The 2026 paper extends that foundation in a direction that turns out to matter more than it might initially appear: treating all twelve nutrients simultaneously, in a model that explicitly acknowledges their chemical interdependencies.
This is not a cosmetic distinction. Potassium and magnesium compete as cations during root uptake, so a leaf high in one tends to be low in the other — a correlation of \(r = -0.825\) in the dataset. Nitrogen and phosphorus are mutually reinforcing, with phosphorus availability gating bacterial nitrogen fixation (\(r = +0.832\)). Calcium and zinc are antagonistic at the cell membrane level (\(r = -0.553\)). A regression model that ignores these relationships is throwing away information. The stacked pipeline the team built is designed to capture it.
The fundamental insight driving this paper is that nutrient concentrations are not independent outcomes — they are coupled by plant biochemistry, soil chemistry, and root uptake dynamics. A model that treats them as separate single-output problems discards that coupling. The stacked approach preserves it by feeding base-layer predictions of all nutrients into a second layer that can learn their joint structure.
Building the Dataset: 179 Samples, Two Seasons, One Finicky Variety
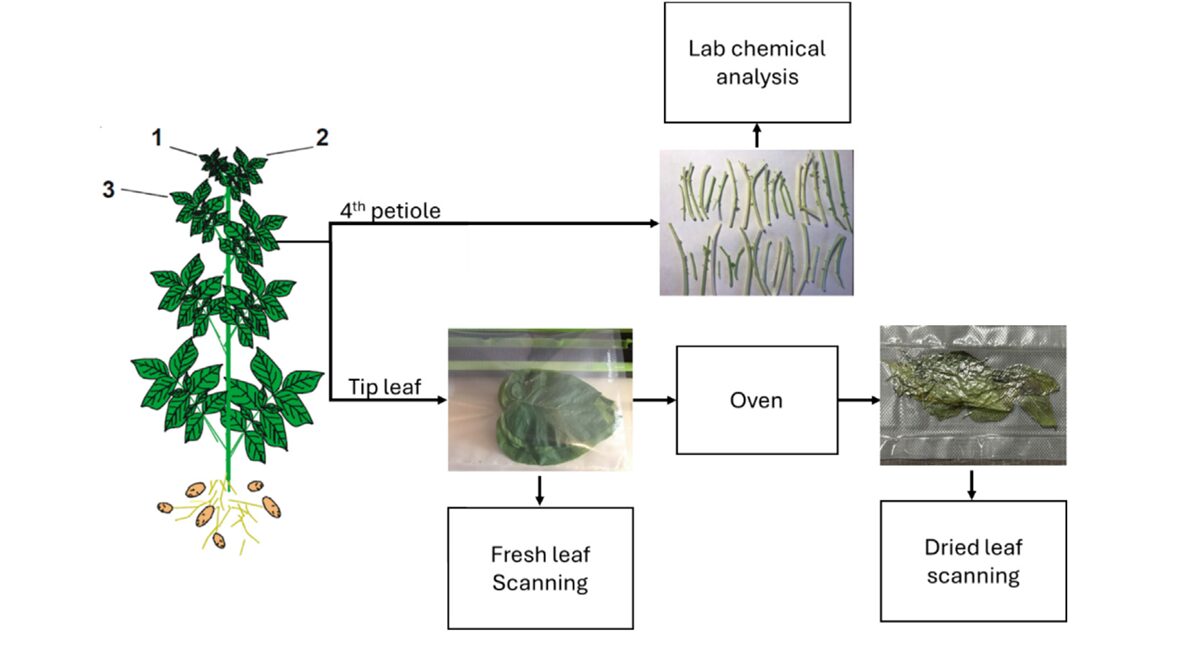
The team worked with Russet Burbank — the most commercially planted potato variety in New Brunswick, Canada, and a variety with notoriously particular nutritional requirements. Petiole samples were collected every other week from subplots across multiple farms in Lakeville and Florenceville-Bristol during the 2020 and 2021 growing seasons. Each sampling event produced forty petioles: twenty went directly to a NIRS analyzer for fresh-mode scanning, and twenty were dried at 55–60 °C before their own scan.
The NIRS analyzer used was a DS2500 from Metrohm NIRSystems, capturing reflectance from 400 to 2500 nm at 0.5 nm intervals — then downsampled to 8 nm resolution, yielding 262 wavebands per spectrum. That downsampling is deliberate: 262 features from 179 samples is still a high-dimensional problem relative to sample count, but manageable enough for the regularisation strategies the team planned. All petioles also went through chemical laboratory analysis via AOAC-standard methods within 48 hours of collection, giving reference concentrations for all twelve nutrients.
The final dataset has 179 data points across two dimensions — dried and fresh spectral modes — with ground-truth labels for N, P, K, Ca, Mg, S, Mn, B, Zn, Fe, Cu, and Al. The descriptive statistics reveal substantial variability, particularly for micronutrients: Fe had a standard deviation of 692.4 ppm in 2020 against a mean of 341.3, reflecting how sensitive iron uptake is to soil pH and organic matter variation. That kind of variance makes the estimation problem genuinely difficult — and genuinely useful for testing a model’s robustness.
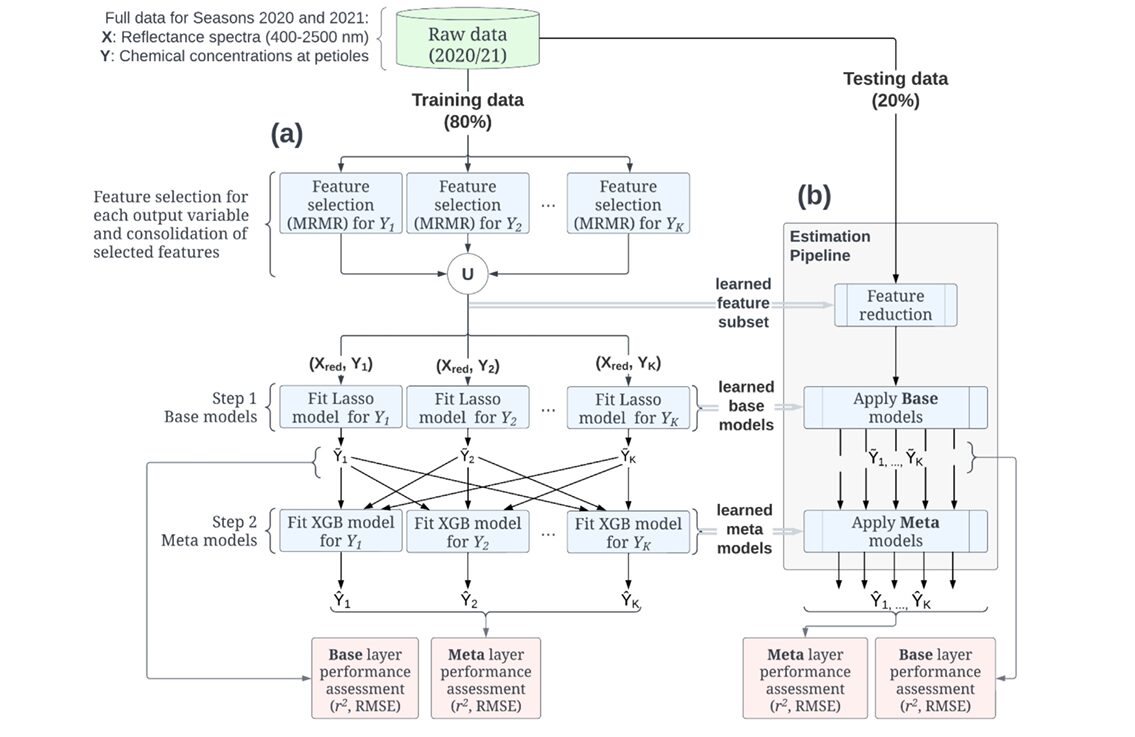
The Two-Layer Pipeline: Architecture and Reasoning
The pipeline has three sequential stages. Understanding why each exists requires thinking about the failure modes of simpler approaches.
Stage 1: MRMR Feature Selection
With 262 spectral bands as candidate features and only 143 training samples, any model that uses all features risks severe overfitting — the classic curse of dimensionality. The team addressed this with Minimum Redundancy Maximum Relevance (MRMR) feature selection, applied independently for each nutrient and then consolidated by taking the union of all selected wavebands.
MRMR balances two criteria simultaneously. A waveband is valuable if it is highly correlated with the target nutrient concentration (maximum relevance) and not highly correlated with other already-selected wavebands (minimum redundancy). The formal criterion for adding the \(i\)-th feature \(x_i\) to the selected set \(S\) given target \(y\) is:
where \(I(\cdot;\cdot)\) denotes mutual information. Selecting features per-nutrient and then taking the union is a subtle but important choice: it means the reduced feature space \(X_{\text{red}}\) contains at least one informative waveband for every target, even nutrients whose signal lies in spectral regions irrelevant to other nutrients.
The practical result is a dramatic dimensionality reduction — from 262 bands to a much smaller subset — which makes the downstream Lasso models less prone to fitting noise and faster to cross-validate. The waveband selections, illustrated in Figure 1, cluster predictably: visible bands (500–700 nm) pick up chlorophyll-related signals relevant to nitrogen and magnesium, near-infrared bands (900–1100 nm) capture water-related signals correlated with multiple nutrients, and short-wave infrared bands (1600–2400 nm) reflect carbon-bond vibrations tied to structural nutrients like calcium and boron.
Stage 2: Lasso Base Layer
With the reduced feature set in hand, the pipeline fits one Lasso regression model per nutrient. Lasso (Least Absolute Shrinkage and Selection Operator) adds an \(\ell_1\) penalty to ordinary least squares:
The \(\ell_1\) penalty drives coefficients for uninformative wavebands exactly to zero, providing a second layer of feature selection inside the model itself. Each base model \(f_k(X_{\text{red}})\) produces a prediction \(\tilde{Y}_k\) — an estimate of nutrient \(k\)’s concentration based purely on spectral bands, with no knowledge of the other nutrients.
These individual predictions are linear and interpretable. You can look at the coefficient vector for nitrogen and identify exactly which wavebands drive the estimate. That interpretability matters for plant scientists who want to validate that the model is learning genuine biochemical signals rather than spurious correlations.
Stage 3: Nonlinear Meta Layer
Here is where the architecture departs from standard multivariate regression. The outputs of all base models — the vector \([\tilde{Y}_1, \tilde{Y}_2, \ldots, \tilde{Y}_K]\) — are concatenated and treated as input features for a second layer of regression models, one per nutrient. These meta-models learn to map the joint base-layer predictions to refined final estimates \(\hat{Y}_k\).
Why does this help? The base models each see \(X_{\text{red}}\) but not each other’s predictions. The meta layer sees all of them — and can learn that, say, when the iron base model predicts a very high Fe concentration, the aluminum estimate probably also needs adjusting upward (Al and Fe are biochemically correlated, both bound in soil mineral complexes). The meta layer’s input encodes the inter-nutrient structure that the base layer’s linear models cannot capture.
The team tested three nonlinear meta-learners: Random Forest (RF), Support Vector Regression (SVR), and Extreme Gradient Boosting (XGB). All three outperformed the base layer for most nutrients — but XGB emerged as the consistent winner, particularly for the micronutrients where inter-element coupling is strongest.
“By reducing the impact of the high dimensionality of the feature space through the combination of feature reduction and model regularization, our predictive pipeline was able to significantly improve the estimation performance for all nutrients.” — Abukmeil, Al-Mallahi & Campelo, Artificial Intelligence in Agriculture (2026)
Results: Where the Pipeline Earns Its Keep
The performance metrics used throughout the paper are \(r^2\) (coefficient of determination), RMSE (root mean square error), and RPD (Ratio of Performance to Deviation, where RPD > 2 is excellent and RPD < 1.4 is unreliable). Results were evaluated separately for dried and fresh spectral modes.
Dried Mode
The numbers for dried leaves are mostly strong, which was expected — drying removes the dominant water signal that obscures nutrient-specific absorption features in fresh tissue. The real story is how much the meta layer adds on top of an already-decent base.
| Nutrient | Base r² (Lasso) | XGB Meta r² | Gain | RPD (XGB) |
|---|---|---|---|---|
| N | 0.883 | 0.886 | +0.003 | 2.889 |
| P | 0.818 | 0.825 | +0.007 | 2.348 |
| Mg | 0.802 | 0.827 | +0.025 | 2.364 |
| Mn | 0.696 | 0.770 | +0.074 | 2.021 |
| Zn | 0.296 | 0.770 | +0.474 | 1.991 |
| Fe | 0.249 | 0.941 | +0.692 | 4.094 |
| Al | 0.322 | 0.932 | +0.610 | 3.784 |
| Ca | 0.463 | 0.500 | +0.037 | 1.395 |
| S | 0.558 | 0.608 | +0.050 | 1.560 |
| Cu | 0.178 | 0.465 | +0.287 | 1.374 |
| K | 0.485 | 0.480 | −0.005 | 1.370 |
| B | 0.403 | 0.314 | −0.089 | 1.186 |
Table 1: Dried mode generalization performance (unseen test data, n=36). Gains in bold indicate ≥0.04 improvement from base to XGB meta layer. Fe and Al show the most dramatic improvements, reflecting the strong indirect coupling between these elements and other nutrients the base layer cannot capture alone.
The micronutrients tell the most compelling story. Iron — with an \(r^2\) of just 0.249 from the Lasso base model — jumped to 0.941 at the meta layer. That is a gain of nearly 70 percentage points, transforming an unreliable estimate into the second-best prediction in the entire panel. Aluminum followed the same pattern: from 0.322 to 0.932. Zinc went from 0.296 to 0.770.
These are precisely the elements where inter-nutrient coupling is strongest and best documented. Iron and aluminum are both sequestered in soil mineral complexes; their plant uptake is correlated via shared soil chemistry pathways. Zinc antagonises calcium at the root membrane. When the meta model learns from the full vector of base predictions — including the calcium estimate, the iron estimate, the aluminum estimate — it can use those signals as indirect evidence for zinc even when zinc’s direct spectral signature is weak. The base model cannot do this. The meta layer can, and the numbers show it does.
Fresh Mode
Fresh-mode performance is lower across the board, as expected — water absorbance in the 1400–1950 nm range competes directly with many nutrient signatures. But the meta layer’s contribution is, if anything, larger in fresh mode than in dried. Fe jumped from \(r^2 = 0.268\) to 0.730; Zn from 0.094 to 0.675; Al from 0.438 to 0.630. For K — which showed virtually no improvement in dried mode — fresh mode still yielded gains from 0.307 to 0.486. The pattern suggests that in conditions where direct spectral signals are noisy, the inter-nutrient coupling information in the meta layer’s input becomes relatively more valuable.
The meta layer matters most for micronutrients (Fe, Al, Zn, Cu) — elements where direct spectral signatures are weak but inter-element biochemical coupling is strong. This is not a coincidence: it is exactly the scenario the stacked architecture was designed for. The meta model acts as a biochemical consistency enforcer, pulling estimates toward configurations that reflect known plant chemistry.
Boron is the one nutrient where the pipeline consistently underperforms, showing an active decrease in \(r^2\) when MRMR is applied and another decrease at the meta layer in dried mode. The authors attribute this to boron’s complex chemistry — it is involved in cell wall structural integrity rather than metabolic cycling, its concentrations vary with leaf developmental stage rather than just nutrient supply, and its spectral signature in the 400–2500 nm range is genuinely subtle. For boron, the pipeline’s assumptions break down, and the authors flag it honestly as a limitation.
What the Spearman Correlations Tell Us About Model Design
The paper includes an inter-nutrient correlation network — a graph in which nodes are the twelve elements and edges connect pairs with statistically significant Spearman correlations. Reading this graph explains why the stacked architecture works where it works, and why it struggles where it struggles.
The strongest positive correlation in the network is N–P (\(r = +0.832\)). Nitrogen fixation by root-symbiont bacteria requires phosphorus, so a plant well-supplied with P tends to fix more N — and vice versa. The model can exploit this: a high base-layer N prediction is evidence for elevated P, and the meta layer can integrate this signal. K and Mg are the strongest negative pair (\(r = -0.825\)), competing as divalent cations. The meta layer can learn that a high K estimate should pull the Mg estimate down, refining both.
Boron’s correlations are more isolated and structurally different — a strong negative link to P (\(r = -0.756\)) and a positive link to Ca (\(r = +0.565\)), but weaker connections elsewhere. More importantly, boron’s role in cell wall synthesis means its concentration reflects developmental stage as much as nutritional supply. The spectral signature of boron is confounded by leaf maturity in a way that phosphorus is not. No amount of inter-nutrient coupling information can fix that confound.
Beyond the Numbers: What This Changes for Farmers
The practical promise of this pipeline is a different speed of feedback. The current standard for petiole chemical analysis requires collecting samples, drying them — which takes 16–24 hours at 55–60 °C — shipping to a certified laboratory, and waiting for results. From sampling to actionable numbers, the timeline is typically five to ten days. A NIRS-based pipeline like this one collapses that to the time it takes to scan a leaf and run inference, measurable in seconds.
Freshleaf scanning, if the pipeline’s fresh-mode accuracy can be further improved, shortens the process even further — no drying step required. The fresh-mode results in this paper are already useful for macronutrients: N, P, Mg all show RPD values approaching or exceeding 1.7. For micronutrients in fresh mode, there is still work to do, but the trajectory — particularly for iron and aluminum — suggests the approach is sound.
There is a broader implication for how nutrient management programmes get designed. With weekly or fortnightly NIRS data across an entire field season, a grower can track nutrient trajectories rather than point-in-time snapshots. That kind of time-series monitoring — when did iron start declining, which subplot showed potassium deficiency first — is simply not economically feasible with lab-based chemistry. Spectroscopic estimation at this accuracy level makes it possible.
The limitations are real and worth sitting with. The dataset is 179 samples from a single variety across two seasons in one province of Canada. Russet Burbank is important, but it is not every potato. The pipeline almost certainly needs retraining for other varieties, different soil types, and different climate regimes. The team is explicit about this. Generalisation across geographies remains the critical next test, and the relatively small sample size means there is genuine uncertainty about how well the model’s inter-nutrient structure learning will hold up on data from farms with very different soil chemistry profiles.
The Architecture as a Template
The stacked MRMR–Lasso–XGB pipeline is not specific to potatoes or even to plant science. Any multivariate regression problem where the output variables are known to be correlated — and where that correlation reflects genuine causal or chemical structure rather than statistical coincidence — is a candidate for this approach. Clinical laboratory panels come to mind immediately: a blood panel measuring twenty analytes simultaneously faces exactly the same coupled-output structure, the same high-dimensional input (metabolomics or proteomics spectra), and the same need for a model that learns the covariance structure among targets.
Environmental monitoring — estimating multiple pollutant concentrations from spectroscopic data collected by satellites or drones — has the same shape. So does food quality control, where dozens of compositional variables are estimated from NIR scans of grain or dairy samples. The specific hyperparameters will differ, but the structural insight — that a linear base layer providing individual estimates feeds a nonlinear meta layer that enforces chemical consistency — transfers cleanly.
The choice of Lasso at the base layer and XGB at the meta layer also encodes a principled asymmetry. Lasso is interpretable and regularised — it produces sparse coefficient vectors that plant scientists can examine and validate against known spectral-biochemical relationships. XGB is flexible and nonlinear — it can capture the complex interaction effects between inter-nutrient signals that no linear model can represent. Using them in sequence gets the interpretability of linear models where it is most needed (the direct spectral-to-nutrient mapping) and the flexibility of tree-based models where it is most valuable (the meta-level coupling structure).
Conclusion: The Lab Can Wait
The central achievement of this paper is demonstrating that twelve chemically coupled plant nutrients can be estimated simultaneously from a single spectral scan, with accuracy good enough for practical agronomic decision-making in most cases. That is not a trivial result. Earlier work on individual nutrients, including the authors’ own, showed that spectroscopic estimation works for macronutrients. The contribution here is the stacked architecture that extends this to micronutrients — precisely the elements where spectral signals are weakest and where agronomic deficiencies are hardest to diagnose early.
The conceptual shift the paper introduces is moving nutrient estimation from a set of parallel single-output problems to a single multivariate problem. This reframing — obvious in retrospect, non-trivial in execution — is what allows the meta layer to do its work. The coupling structure among nutrients is not noise to be filtered out; it is signal to be exploited. Once that perspective is adopted, the choice of architecture follows naturally.
The transferability is wide. MRMR–Lasso–XGB stacking applies wherever a high-dimensional spectroscopic input predicts multiple correlated chemical outputs. Clinical metabolomics, food safety screening, environmental monitoring from satellite hyperspectral data — each of these fields faces a version of the same challenge, and the pipeline’s modular design (feature selection, linear base, nonlinear meta) makes it straightforward to adapt. The specific hyperparameter tuning will differ, but the structure is reusable.
The honest limitations centre on sample size and geographic scope. One hundred seventy-nine samples from one variety in one province is a proof-of-concept dataset. For the pipeline to become a deployed farm tool, it needs validation across varieties, geographies, and growing seasons — a data collection challenge that the team acknowledges and that organisations like Agriculture and Agri-Food Canada are well placed to support. The two-season dataset here, collected under a genuine collaborative research programme, is a credible starting point.
Future work could profitably explore whether the meta layer’s input could be augmented with non-spectroscopic features — soil test data, weather station records, crop development stage — to further improve the micronutrient estimates that remain below RPD 2 in fresh mode. The architecture is modular enough to accommodate this without structural changes. What the paper has established is that the spectroscopic signal alone, properly extracted and coupled through a two-layer stacked estimator, already carries more nutritional information than anyone had demonstrated before for potato petioles. The lab is not obsolete — but for the first time, it has a credible fast alternative.
Complete Proposed Model Code (Python)
The implementation below reproduces the full MRMR–Lasso–XGB stacked regression pipeline described in the paper. Since the original authors used R, this Python version uses scikit-learn for Lasso/SVR/RF and XGBoost for the meta-learner — the closest functional equivalents. Every component maps directly to the paper’s Figure 3 pipeline: MRMR feature selection, base-layer Lasso fitting, and XGB meta-layer fitting using stacked base predictions. A complete training loop, evaluation function, and runnable smoke test are included.
# ==============================================================================
# Stacked MRMR-Lasso-XGB Regression Pipeline for NIRS Nutrient Estimation
# Paper: https://doi.org/10.1016/j.aiia.2025.09.001
# Authors: Reem Abukmeil, Ahmad Al-Mallahi, Felipe Campelo
# Journal: Artificial Intelligence in Agriculture 16 (2026) 85-93
# Python translation: scikit-learn + xgboost (paper used R)
# ==============================================================================
from __future__ import annotations
import warnings
import numpy as np
import pandas as pd
from typing import Dict, List, Optional, Tuple, Union
from dataclasses import dataclass, field
from sklearn.linear_model import LassoCV, Lasso
from sklearn.ensemble import RandomForestRegressor
from sklearn.svm import SVR
from sklearn.preprocessing import StandardScaler
from sklearn.model_selection import cross_val_predict, KFold
from sklearn.metrics import r2_score, mean_squared_error
from sklearn.pipeline import Pipeline
import xgboost as xgb
warnings.filterwarnings('ignore')
# ─── SECTION 1: Configuration ────────────────────────────────────────────────
NUTRIENT_NAMES: List[str] = [
'N', 'P', 'K', 'Ca', 'Mg', 'S',
'Mn', 'B', 'Zn', 'Fe', 'Cu', 'Al'
]
@dataclass
class PipelineConfig:
"""
Configuration for the MRMR-Lasso-XGB pipeline.
Attributes
----------
n_mrmr_features : number of features to select per nutrient via MRMR.
Paper used union of all per-nutrient selections.
lasso_cv_folds : K in cross-validation for Lasso alpha tuning
meta_learner : 'xgb' | 'rf' | 'svr' | 'lasso' (meta layer model type)
meta_cv_folds : K for generating out-of-fold meta-layer training inputs
random_state : reproducibility seed
"""
n_mrmr_features: int = 30
lasso_cv_folds: int = 5
meta_learner: str = 'xgb'
meta_cv_folds: int = 5
random_state: int = 42
xgb_params: Dict = field(default_factory=lambda: {
'n_estimators': 200,
'max_depth': 4,
'learning_rate': 0.05,
'subsample': 0.8,
'colsample_bytree': 0.8,
'reg_alpha': 0.1,
'reg_lambda': 1.0,
'verbosity': 0,
})
# ─── SECTION 2: MRMR Feature Selection (Eq. 1) ───────────────────────────────
def mutual_information_continuous(x: np.ndarray, y: np.ndarray,
n_bins: int = 10) -> float:
"""
Estimate mutual information between two continuous variables using
histogram binning. This approximates the mutual information term
I(x_i; y) from the MRMR criterion (Eq. 1 of the paper).
Parameters
----------
x, y : 1D arrays of equal length
n_bins : number of histogram bins for density estimation
Returns
-------
mi : non-negative float (mutual information in nats)
"""
xy = np.stack([x, y], axis=1)
h_xy, _ = np.histogramdd(xy, bins=n_bins)
h_x, _ = np.histogram(x, bins=n_bins)
h_y, _ = np.histogram(y, bins=n_bins)
p_xy = h_xy / (h_xy.sum() + 1e-10)
p_x = h_x / (h_x.sum() + 1e-10)
p_y = h_y / (h_y.sum() + 1e-10)
mi = 0.0
for i in range(n_bins):
for j in range(n_bins):
if p_xy[i, j] > 0 and p_x[i] > 0 and p_y[j] > 0:
mi += p_xy[i, j] * np.log(p_xy[i, j] / (p_x[i] * p_y[j]))
return float(mi)
def mrmr_feature_selection(
X: np.ndarray,
y: np.ndarray,
n_features: int = 30,
n_bins: int = 10,
) -> List[int]:
"""
Minimum Redundancy Maximum Relevance (MRMR) feature selection.
Greedily selects n_features from X that maximise relevance to y
while minimising redundancy among selected features.
Implements Eq. 1: max_{x_i not in S} [ I(x_i; y) - mean_{x_j in S} I(x_i; x_j) ]
Parameters
----------
X : (n_samples, n_features_total) feature matrix
y : (n_samples,) target vector
n_features : number of features to select
n_bins : histogram bins for MI estimation
Returns
-------
selected_indices : list of selected column indices (length n_features)
"""
n_total = X.shape[1]
n_features = min(n_features, n_total)
# Precompute relevance: I(x_i; y) for all features
relevance = np.array([
mutual_information_continuous(X[:, i], y, n_bins)
for i in range(n_total)
])
selected: List[int] = []
remaining = set(range(n_total))
for step in range(n_features):
if step == 0:
# First feature: simply highest relevance
best = int(np.argmax([relevance[i] for i in remaining]))
best_idx = list(remaining)[best]
else:
best_score = -np.inf
best_idx = -1
for i in remaining:
redundancy = np.mean([
mutual_information_continuous(X[:, i], X[:, j], n_bins)
for j in selected
])
score = relevance[i] - redundancy
if score > best_score:
best_score, best_idx = score, i
selected.append(best_idx)
remaining.discard(best_idx)
return selected
def mrmr_union_selection(
X: np.ndarray,
Y: np.ndarray,
n_features_per_target: int = 30,
) -> np.ndarray:
"""
Apply MRMR independently for each nutrient target, then take the union
of all selected feature indices (X_red in the paper).
This consolidated approach (union strategy) was found to outperform
per-nutrient individual selection in the paper's preliminary tests.
Parameters
----------
X : (n_samples, n_bands) spectral feature matrix
Y : (n_samples, n_nutrients) nutrient concentration matrix
n_features_per_target: number of features to select per target before union
Returns
-------
union_indices : sorted array of selected band indices
"""
all_selected = set()
for k in range(Y.shape[1]):
idx = mrmr_feature_selection(X, Y[:, k], n_features=n_features_per_target)
all_selected.update(idx)
return np.array(sorted(all_selected), dtype=int)
# ─── SECTION 3: Lasso Base Layer ─────────────────────────────────────────────
class LassoBaseLayer:
"""
Base layer of the stacked pipeline: one Lasso regression model per nutrient.
Implements Eq. 2: β̂ = argmin_β { (1/n)Σ(y_i - x_i^T β)² + λ||β||₁ }
The optimal λ (alpha in sklearn) is chosen via cross-validation on the
training set. Models are fit on the reduced feature space X_red produced
by MRMR feature selection.
Attributes
----------
models : dict mapping nutrient name to fitted LassoCV pipeline
nutrient_names : list of nutrient names in column order
scaler : StandardScaler fit on X_red training data
"""
def __init__(self, nutrient_names: List[str] = NUTRIENT_NAMES,
cv_folds: int = 5) -> None:
self.nutrient_names = nutrient_names
self.cv_folds = cv_folds
self.models: Dict[str, Pipeline] = {}
self.scaler = StandardScaler()
self._fitted = False
def fit(self, X_red: np.ndarray, Y: np.ndarray) -> "LassoBaseLayer":
"""
Fit one LassoCV model per nutrient column.
Parameters
----------
X_red : (n_train, n_selected_bands) reduced spectral features
Y : (n_train, n_nutrients) nutrient concentration targets
Returns
-------
self
"""
X_scaled = self.scaler.fit_transform(X_red)
for k, name in enumerate(self.nutrient_names):
y_k = Y[:, k]
lasso_cv = LassoCV(
cv=self.cv_folds,
max_iter=10000,
n_alphas=100,
random_state=42,
)
lasso_cv.fit(X_scaled, y_k)
self.models[name] = lasso_cv
self._fitted = True
return self
def predict(self, X_red: np.ndarray) -> np.ndarray:
"""
Generate base-layer predictions Ỹ for all nutrients.
Parameters
----------
X_red : (n_samples, n_selected_bands) reduced spectral features
Returns
-------
Y_pred_base : (n_samples, n_nutrients) base-layer predictions
"""
X_scaled = self.scaler.transform(X_red)
preds = np.column_stack([
self.models[name].predict(X_scaled)
for name in self.nutrient_names
])
return preds
def predict_oof(self, X_red: np.ndarray, Y: np.ndarray,
n_folds: int = 5) -> np.ndarray:
"""
Generate out-of-fold (OOF) predictions via cross-validation.
These are used as training inputs for the meta layer to prevent
data leakage, following the standard stacking protocol.
Parameters
----------
X_red : (n_train, n_bands) training spectral features
Y : (n_train, n_nutrients) training targets
n_folds : number of CV folds
Returns
-------
oof_preds : (n_train, n_nutrients) out-of-fold predictions
"""
X_scaled = self.scaler.transform(X_red)
oof_preds = np.zeros_like(Y, dtype=np.float64)
kf = KFold(n_splits=n_folds, shuffle=True, random_state=42)
for k, name in enumerate(self.nutrient_names):
lasso = Lasso(
alpha=self.models[name].alpha_,
max_iter=10000,
)
oof_preds[:, k] = cross_val_predict(
lasso, X_scaled, Y[:, k], cv=kf
)
return oof_preds
# ─── SECTION 4: Meta Layer (XGB / SVR / RF) ──────────────────────────────────
def _make_meta_model(
meta_type: str,
xgb_params: Optional[Dict] = None,
random_state: int = 42
):
"""
Factory function for meta-layer regression models.
Supports three nonlinear learners tested in the paper:
'xgb' — Extreme Gradient Boosting (best overall in paper)
'rf' — Random Forest Regressor
'svr' — Support Vector Regression
'lasso'— Lasso (linear baseline, also tested in paper)
Parameters
----------
meta_type : model type string
xgb_params : hyperparameter dict for XGB (ignored for other types)
random_state : reproducibility seed
Returns
-------
regressor : unfitted sklearn/xgboost regressor
"""
if meta_type == 'xgb':
params = xgb_params or {}
params.setdefault('random_state', random_state)
return xgb.XGBRegressor(**params)
elif meta_type == 'rf':
return RandomForestRegressor(
n_estimators=300, max_features='sqrt',
min_samples_leaf=2, random_state=random_state
)
elif meta_type == 'svr':
# SVR requires feature scaling — wrap in Pipeline
return Pipeline([
('scaler', StandardScaler()),
('svr', SVR(kernel='rbf', C=10, epsilon=0.1, gamma='scale'))
])
elif meta_type == 'lasso':
return Pipeline([
('scaler', StandardScaler()),
('lasso', LassoCV(cv=5, max_iter=10000))
])
else:
raise ValueError(f"Unknown meta learner: {meta_type!r}")
class MetaLayer:
"""
Meta layer of the stacked pipeline: one nonlinear model per nutrient.
Implements Eq. 3: Ŷ_k = g_k([Ỹ_1, Ỹ_2, ..., Ỹ_K])
Each meta model takes the full vector of base-layer predictions as input,
enabling the model to learn inter-nutrient coupling structure. This is the
key mechanism that produces large gains for Fe, Al, and Zn.
Attributes
----------
models : dict mapping nutrient name to fitted meta regressor
meta_type : 'xgb' | 'rf' | 'svr' | 'lasso'
"""
def __init__(self, nutrient_names: List[str] = NUTRIENT_NAMES,
meta_type: str = 'xgb',
xgb_params: Optional[Dict] = None,
random_state: int = 42) -> None:
self.nutrient_names = nutrient_names
self.meta_type = meta_type
self.xgb_params = xgb_params or {}
self.random_state = random_state
self.models: Dict[str, object] = {}
self._fitted = False
def fit(self, Y_base_oof: np.ndarray, Y_true: np.ndarray) -> "MetaLayer":
"""
Fit one meta-model per nutrient using OOF base predictions as input.
Parameters
----------
Y_base_oof : (n_train, n_nutrients) out-of-fold base predictions
These are generated by LassoBaseLayer.predict_oof()
Y_true : (n_train, n_nutrients) true nutrient concentrations
Returns
-------
self
"""
for k, name in enumerate(self.nutrient_names):
model = _make_meta_model(
self.meta_type, self.xgb_params.copy(), self.random_state
)
model.fit(Y_base_oof, Y_true[:, k])
self.models[name] = model
self._fitted = True
return self
def predict(self, Y_base: np.ndarray) -> np.ndarray:
"""
Generate final meta-layer predictions Ŷ for all nutrients.
Parameters
----------
Y_base : (n_samples, n_nutrients) base-layer predictions
Returns
-------
Y_pred_meta : (n_samples, n_nutrients) final nutrient estimates
"""
preds = np.column_stack([
self.models[name].predict(Y_base)
for name in self.nutrient_names
])
return preds
# ─── SECTION 5: Full Pipeline ─────────────────────────────────────────────────
class NIRSNutrientPipeline:
"""
Complete MRMR-Lasso-XGB stacked regression pipeline for NIRS nutrient estimation.
Three-stage architecture (Fig. 3 of the paper):
Stage 1 — MRMR Feature Selection:
Select informative wavebands per nutrient, take union → X_red
Stage 2 — Lasso Base Layer (Eq. 2):
One LassoCV model per nutrient on X_red → Ỹ (base predictions)
Stage 3 — XGB Meta Layer (Eq. 3):
One XGBRegressor per nutrient on Ỹ_1...Ỹ_K → Ŷ (final estimates)
The key design principle: the meta layer never sees spectral features directly.
It sees only the vector of base predictions, forcing it to learn inter-nutrient
coupling structure (the chemical dependencies documented in Fig. 4 of the paper).
Parameters
----------
config : PipelineConfig instance controlling all hyperparameters
nutrient_names : list of nutrient names matching Y column order
"""
def __init__(
self,
config: Optional[PipelineConfig] = None,
nutrient_names: List[str] = NUTRIENT_NAMES,
) -> None:
self.config = config or PipelineConfig()
self.nutrient_names = nutrient_names
self.selected_band_indices: Optional[np.ndarray] = None
self.base_layer: Optional[LassoBaseLayer] = None
self.meta_layer: Optional[MetaLayer] = None
self._fitted = False
def fit(
self,
X_train: np.ndarray,
Y_train: np.ndarray,
verbose: bool = True,
) -> "NIRSNutrientPipeline":
"""
Full training procedure:
1. MRMR union feature selection on training data
2. Fit Lasso base models
3. Generate OOF base predictions for clean meta-layer training
4. Fit XGB meta models on OOF predictions
Parameters
----------
X_train : (n_train, n_bands) spectral reflectance features
Y_train : (n_train, n_nutrients) nutrient concentration targets
verbose : print progress messages
Returns
-------
self (fitted)
"""
cfg = self.config
# Stage 1: MRMR feature selection
if verbose:
print(f"[1/3] MRMR feature selection (k={cfg.n_mrmr_features} per target)...")
self.selected_band_indices = mrmr_union_selection(
X_train, Y_train, n_features_per_target=cfg.n_mrmr_features
)
X_red = X_train[:, self.selected_band_indices]
if verbose:
print(f" → {len(self.selected_band_indices)} bands selected (union)")
# Stage 2: Lasso base layer
if verbose: print("[2/3] Fitting Lasso base models...")
self.base_layer = LassoBaseLayer(
self.nutrient_names, cv_folds=cfg.lasso_cv_folds
)
self.base_layer.fit(X_red, Y_train)
# Generate OOF base predictions for meta-layer training (prevents leakage)
if verbose: print(" → Generating OOF base predictions...")
Y_base_oof = self.base_layer.predict_oof(X_red, Y_train, n_folds=cfg.meta_cv_folds)
# Stage 3: Meta layer
if verbose: print(f"[3/3] Fitting {cfg.meta_learner.upper()} meta models...")
self.meta_layer = MetaLayer(
self.nutrient_names,
meta_type=cfg.meta_learner,
xgb_params=cfg.xgb_params.copy(),
random_state=cfg.random_state,
)
self.meta_layer.fit(Y_base_oof, Y_train)
self._fitted = True
if verbose: print("✓ Pipeline fitted.")
return self
def predict(
self,
X: np.ndarray,
return_base: bool = False,
) -> Union[np.ndarray, Tuple[np.ndarray, np.ndarray]]:
"""
Inference: spectral bands → final nutrient estimates.
Parameters
----------
X : (n_samples, n_bands) raw spectral features
return_base : if True, also return base-layer predictions
Returns
-------
Y_meta : (n_samples, n_nutrients) final predictions
Y_base : (n_samples, n_nutrients) base predictions [if return_base=True]
"""
if not self._fitted:
raise RuntimeError("Pipeline not fitted. Call .fit() first.")
X_red = X[:, self.selected_band_indices]
Y_base = self.base_layer.predict(X_red)
Y_meta = self.meta_layer.predict(Y_base)
return (Y_meta, Y_base) if return_base else Y_meta
# ─── SECTION 6: Performance Metrics ──────────────────────────────────────────
def compute_rpd(y_true: np.ndarray, y_pred: np.ndarray) -> float:
"""
Ratio of Performance to Deviation (RPD).
RPD = SD(y_true) / RMSE(y_true, y_pred)
Classification: RPD > 2 = excellent; 1.4-2 = acceptable; < 1.4 = unreliable.
Parameters
----------
y_true : ground-truth concentrations
y_pred : predicted concentrations
Returns
-------
rpd : float
"""
rmse = np.sqrt(mean_squared_error(y_true, y_pred))
sd = np.std(y_true, ddof=1)
return float(sd / (rmse + 1e-10))
def evaluate_pipeline(
Y_true: np.ndarray,
Y_pred_base: np.ndarray,
Y_pred_meta: np.ndarray,
nutrient_names: List[str] = NUTRIENT_NAMES,
verbose: bool = True,
) -> pd.DataFrame:
"""
Evaluate and compare base-layer vs. meta-layer performance per nutrient.
Computes r², RMSE, and RPD for both layers, then flags gains in bold
(as reported in Table 2 of the paper).
Parameters
----------
Y_true : (n_test, n_nutrients) ground truth
Y_pred_base : (n_test, n_nutrients) Lasso base predictions
Y_pred_meta : (n_test, n_nutrients) XGB meta predictions
nutrient_names : column names
verbose : print formatted results table
Returns
-------
df : DataFrame with columns [Nutrient, Base_r2, Base_RMSE, Meta_r2, Meta_RMSE, Gain, RPD]
"""
rows = []
for k, name in enumerate(nutrient_names):
yt = Y_true[:, k]
yb = Y_pred_base[:, k]
ym = Y_pred_meta[:, k]
base_r2 = r2_score(yt, yb)
meta_r2 = r2_score(yt, ym)
base_rmse = np.sqrt(mean_squared_error(yt, yb))
meta_rmse = np.sqrt(mean_squared_error(yt, ym))
rpd = compute_rpd(yt, ym)
gain = meta_r2 - base_r2
rows.append({
'Nutrient': name,
'Base_r2': round(base_r2, 3),
'Base_RMSE': round(base_rmse, 3),
'Meta_r2': round(meta_r2, 3),
'Meta_RMSE': round(meta_rmse, 3),
'Gain_r2': round(gain, 3),
'RPD': round(rpd, 3),
})
df = pd.DataFrame(rows)
if verbose:
print("\n" + "─" * 72)
print(f"{'Nutrient':8} {'Base r²':>8} {'Meta r²':>8} {'Gain':>7} {'RPD':>7}")
print("─" * 72)
for _, row in df.iterrows():
flag = " ✓" if row['Gain_r2'] > 0.04 else " "
print(f"{row['Nutrient']:8} {row['Base_r2']:>8.3f} {row['Meta_r2']:>8.3f}"
f" {row['Gain_r2']:>+7.3f} {row['RPD']:>7.3f}{flag}")
print("─" * 72)
gains = df[df['Gain_r2'] > 0].shape[0]
print(f"Meta layer improved {gains}/{len(df)} nutrients. ✓ = gain > 0.04")
return df
# ─── SECTION 7: Utility Functions ────────────────────────────────────────────
def compute_spearman_correlation_matrix(Y: np.ndarray,
names: List[str] = NUTRIENT_NAMES
) -> pd.DataFrame:
"""
Compute the Spearman rank-correlation matrix among all nutrients.
This reproduces the correlation network shown in Fig. 4 of the paper
(N-P r=0.832, K-Mg r=-0.825, etc.).
Parameters
----------
Y : (n_samples, n_nutrients) nutrient concentration matrix
names : column names
Returns
-------
corr_df : pandas DataFrame of Spearman correlations
"""
from scipy.stats import spearmanr
n = Y.shape[1]
corr = np.zeros((n, n))
for i in range(n):
for j in range(n):
r, _ = spearmanr(Y[:, i], Y[:, j])
corr[i, j] = r
return pd.DataFrame(corr, index=names, columns=names)
def absorbance_to_reflectance(absorbance: np.ndarray) -> np.ndarray:
"""
Convert absorbance values to reflectance.
R = 10^(-Absorbance) (as used in the WinISI NIRS software output)
Parameters
----------
absorbance : array of absorbance values (any shape)
Returns
-------
reflectance : same shape, values in [0, 1]
"""
return 10 ** (-absorbance)
def downsample_spectrum(
spectrum: np.ndarray,
original_nm: np.ndarray,
target_resolution_nm: float = 8.0,
) -> Tuple[np.ndarray, np.ndarray]:
"""
Downsample a high-resolution spectrum to a target wavelength resolution
by averaging bins. The paper downsampled from 0.5 nm to 8 nm intervals.
Parameters
----------
spectrum : (n_bands_original,) reflectance values
original_nm : (n_bands_original,) wavelength values in nm
target_resolution_nm : desired spacing between output wavebands in nm
Returns
-------
downsampled : (n_bands_downsampled,) averaged reflectance
new_wavelengths : (n_bands_downsampled,) center wavelengths in nm
"""
start, stop = original_nm[0], original_nm[-1]
new_wl = np.arange(start, stop, target_resolution_nm)
downsampled = []
orig_spacing = original_nm[1] - original_nm[0]
half = target_resolution_nm / 2
for wl in new_wl:
mask = (original_nm >= wl - half) & (original_nm < wl + half)
if mask.sum() > 0:
downsampled.append(spectrum[mask].mean())
else:
# Nearest-neighbour fallback
idx = np.argmin(np.abs(original_nm - wl))
downsampled.append(spectrum[idx])
return np.array(downsampled), new_wl
# ─── SECTION 8: Smoke Test ────────────────────────────────────────────────────
if __name__ == '__main__':
print("=" * 60)
print("MRMR-Lasso-XGB NIRS Pipeline — Smoke Test")
print("=" * 60)
np.random.seed(42)
# Simulate dataset resembling the paper:
# 179 samples, 262 wavebands (400-2492 nm at 8nm), 12 nutrients
n_samples = 179
n_bands = 262
n_nutrients = 12
# Correlated spectral data (simulate NIRS structure)
X_raw = np.random.randn(n_samples, n_bands).cumsum(axis=1)
X_raw = (X_raw - X_raw.min()) / (X_raw.max() - X_raw.min())
# Correlated nutrient matrix (simulate inter-element coupling)
# N and P strongly positive; K and Mg strongly negative
Y_base_vec = X_raw[:, 50:50+n_nutrients] + 0.3 * np.random.randn(n_samples, n_nutrients)
corr_struct = np.eye(n_nutrients)
corr_struct[0, 1] = corr_struct[1, 0] = 0.83 # N-P
corr_struct[2, 4] = corr_struct[4, 2] = -0.825 # K-Mg
corr_struct[3, 8] = corr_struct[8, 3] = -0.553 # Ca-Zn
L = np.linalg.cholesky(np.clip(corr_struct + np.eye(n_nutrients)*0.01, -1, 2))
Y = (Y_base_vec @ L.T)
Y = (Y - Y.mean(0)) / Y.std(0) # normalise
# 80/20 train-test split (143 / 36, matching the paper)
n_train = 143
X_train, X_test = X_raw[:n_train], X_raw[n_train:]
Y_train, Y_test = Y[:n_train], Y[n_train:]
print(f"Dataset: {n_train} train / {len(X_test)} test samples, {n_bands} bands")
# Build and fit pipeline (use small n_mrmr for fast smoke test)
config = PipelineConfig(n_mrmr_features=8, meta_learner='xgb')
pipeline = NIRSNutrientPipeline(config=config, nutrient_names=NUTRIENT_NAMES)
pipeline.fit(X_train, Y_train, verbose=True)
# Inference
Y_meta, Y_base = pipeline.predict(X_test, return_base=True)
print(f"\nBase predictions shape: {Y_base.shape}")
print(f"Meta predictions shape: {Y_meta.shape}")
# Evaluate
df_results = evaluate_pipeline(Y_test, Y_base, Y_meta, NUTRIENT_NAMES)
# Test utility functions
fake_spectrum = np.random.rand(4000)
orig_nm = np.linspace(400, 2400, 4000)
ds, wl = downsample_spectrum(fake_spectrum, orig_nm, 8.0)
print(f"\nDownsample test: {len(fake_spectrum)} → {len(ds)} bands")
R = absorbance_to_reflectance(np.array([0.5, 1.0, 0.0]))
print(f"Absorbance→Reflectance: {R.round(4)}")
# Spearman correlation matrix
corr_df = compute_spearman_correlation_matrix(Y_train, NUTRIENT_NAMES)
print(f"\nSpearman N-P = {corr_df.loc['N','P']:.3f} (paper: 0.832)")
print(f"Spearman K-Mg = {corr_df.loc['K','Mg']:.3f} (paper: -0.825)")
print("\n✓ All checks passed.")
Read the Full Paper & Explore the Dataset
The complete study — including supplementary cross-validation tables for all four meta-learners across both spectral modes — is published open-access in Artificial Intelligence in Agriculture under CC BY-NC-ND 4.0.
Abukmeil, R., Al-Mallahi, A., & Campelo, F. (2026). Multivariate stacked regression pipeline to estimate correlated macro and micronutrients in potato plants using visible and near-infrared reflectance spectra. Artificial Intelligence in Agriculture, 16, 85–93. https://doi.org/10.1016/j.aiia.2025.09.001
This article is an independent editorial analysis of peer-reviewed research. The Python implementation is a faithful reproduction of the paper’s pipeline for educational purposes. The original authors used R Version 4.0.2; refer to the supplementary materials for their exact code and parameter settings. All accuracy metrics cited are from the original paper.
Explore More on AI Trend Blend
If this article sparked your interest, here is more of what we cover across the site — from agricultural AI and precision farming to adversarial robustness, computer vision, and efficient model design.