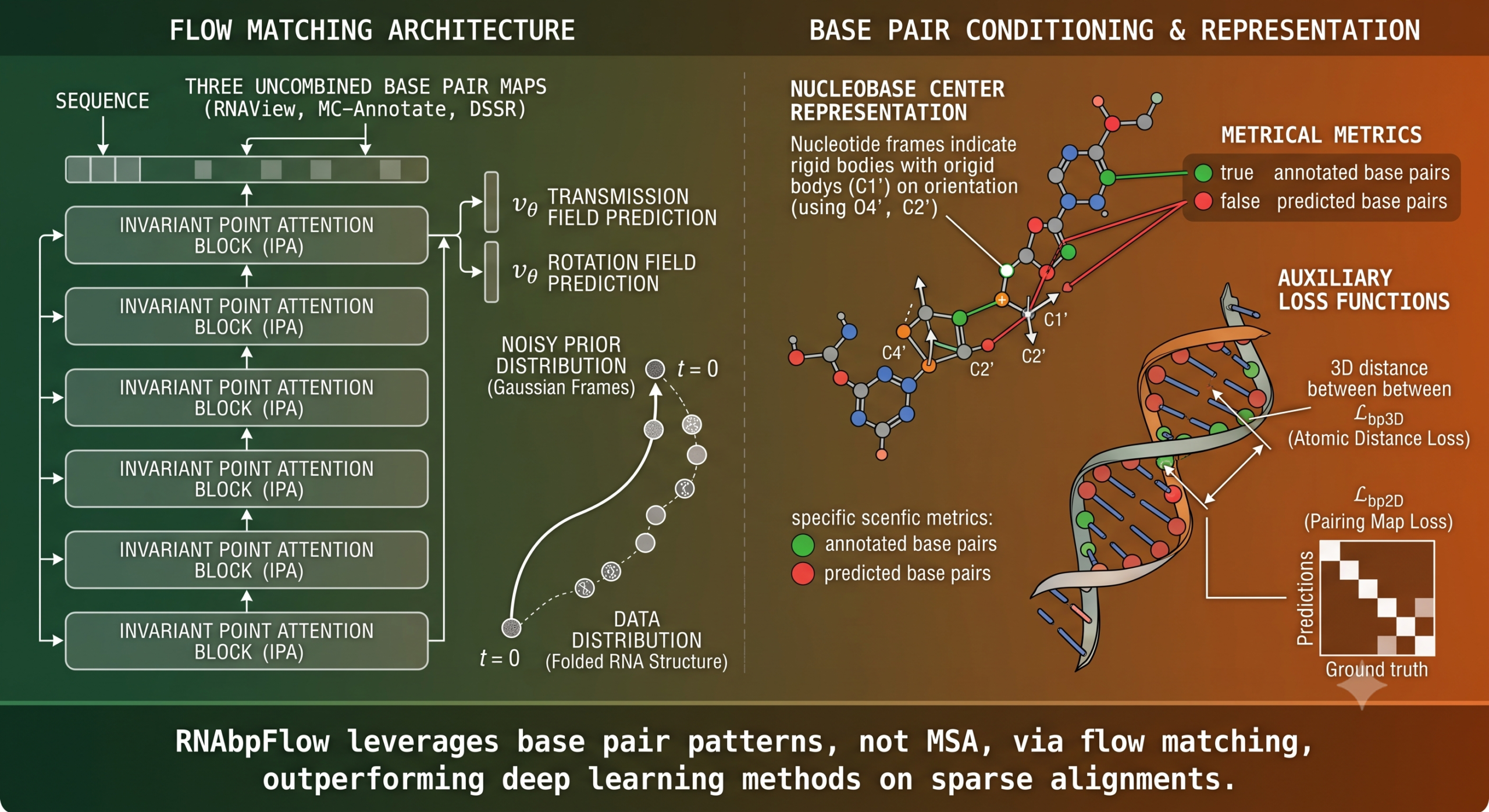
- RNAbpFlow is an SE(3) equivariant flow matching model that generates all atom RNA 3D structures directly from sequence and base pairing information, with no multiple sequence alignment and no homologous template required.
- Base pairs are extracted with three separate annotation tools and fed in as three uncombined binary maps, which lets the model absorb canonical and noncanonical pairing patterns that any single tool would miss on its own.
- Two new base pair centric loss terms, one operating on 3D atomic distance and one on the 2D pairing map, push the model to actually honor the pairing pattern it was given rather than treating it as a soft suggestion.
- Against a molecular dynamics sampling method called RNAJP, RNAbpFlow reaches a mean lDDT of 0.66 against 0.59 and a mean TM-score of 0.38 against 0.32 across 12 benchmark RNAs.
- On the CASP16 blind test, RNAbpFlow beats both top ranked automated servers, AF3-server and Yang-Server, despite those servers having access to deep evolutionary alignments that RNAbpFlow never sees.
- The model’s biggest remaining weakness is that it depends heavily on how good its input base pairs are, and predicted base pairs get noticeably less reliable as RNA sequences get longer than 200 nucleotides.
Why RNA never got its AlphaFold moment the easy way
Interest in RNA structure has been climbing for a reason that has nothing to do with academic curiosity. RNA based therapeutics are now a real category of medicine, and designing an RNA molecule with a specific function starts with knowing what shape it actually folds into. Experimental methods including X ray crystallography, nuclear magnetic resonance spectroscopy and cryo electron microscopy can answer that question, but they are slow, expensive and often unable to keep pace with how flexible RNA molecules really are. A computational method that can fill that gap has obvious value, both for understanding how RNA does what it does inside a cell and for accelerating the design of new RNA drugs.
The traditional computational toolkit split into two camps. Template based approaches such as ModeRNA and RNAbuilder lean on structures that already resemble the target, which only works if something similar has already been solved experimentally. Physics and knowledge based methods such as FARFAR2, 3dRNA, RNAComposer and Vfold3D instead assemble a structure out of prebuilt fragments guided by biophysical potentials, an approach that has done real work in community challenges such as RNA Puzzles and CASP, but one that becomes computationally punishing once the target RNA gets large or its topology gets complicated. Both camps are bottlenecked in the same place, by how little experimentally solved RNA structure actually exists in the Protein Data Bank compared with proteins.
Deep learning looked like the obvious way out, and a wave of methods including DRfold, trRosettaRNA, trRosettaRNA2, RoseTTAFoldNA, RhoFold+ and NuFold arrived to attack RNA structure with attention based architectures directly inspired by AlphaFold 2’s success on proteins. Almost all of them, DRfold being the notable exception, still lean on evolutionary sequence information, either explicitly through multiple sequence alignments or implicitly through a biological language model trained on large sequence corpora. That dependency is where RNA quietly punishes methods borrowed from the protein world. Building a useful alignment for an RNA sequence is genuinely hard, because base pairing is isosteric, meaning very different sequences can form geometrically equivalent pairs, which actively works against standard sequence alignment logic. Many of these methods also underuse the RNA base pairing pattern itself, even though that 2D pattern is one of the strongest known determinants of the eventual 3D fold. And every one of them, evolutionary information or not, still outputs a single static structure, which sits uneasily with the fact that RNA molecules routinely exist as a shifting population of conformations rather than one fixed shape.
The paper’s framing is refreshingly specific about what RNA needs that protein methods do not automatically provide. Evolutionary alignments are hard to build for RNA, and base pairing carries more of the 3D signal than sequence homology does. A method built around base pairing rather than alignment depth is playing to RNA’s actual statistical strengths instead of importing a protein shaped solution and hoping it transfers.
What RNAbpFlow actually does differently
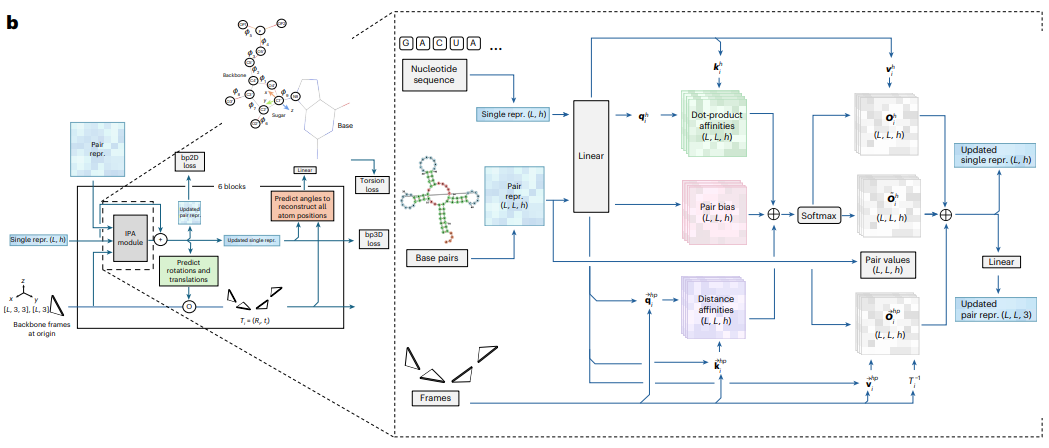
RNAbpFlow sits on the shoulders of FrameFlow, a flow matching formulation originally built for fast protein backbone generation using the SE(3) frame representation, which describes each residue as a rigid body with a position and an orientation rather than as a cloud of independent atoms. RNAbpFlow adapts that idea to RNA using the nucleobase center representation introduced in NuFold, representing each nucleotide as a frame built from the Cartesian coordinates of its C1 prime atom as the local origin, with O4 prime, C1 prime and C2 prime defining the orientation. Starting from a full sequence of these frames sampled from Gaussian noise, the model iteratively refines them toward a real folded structure while conditioning the whole process on two things at once, the nucleotide sequence and the base pairing pattern.
The paper describes three specific methodological contributions, and each one earns its place in the final ablation numbers rather than sitting there as a nice idea that turned out not to matter.
First, base pairing information comes from three separate, independently run annotation tools, RNAView, MC-Annotate and DSSR, and all three resulting binary contact maps are fed into the model as separate channels rather than merged into one consensus map. Any single annotation tool applies its own geometric rules for what counts as a pair, which means the three tools disagree with each other on plenty of edge cases. Rather than trying to reconcile that disagreement before training, RNAbpFlow just hands the model all three views and lets it learn what to do with the discrepancy.
Second, the nucleobase center representation lets the model reconstruct every atom of the molecule directly, including the full set of rotatable bonds in each base, without a separate geometry cleanup step afterward. The three base frame atoms come straight from the learned frame, the first ring nitrogen is placed using simple tetrahedral geometry, and the remaining atoms are organized into ten additional frames whose positions come from nine predicted torsion angles. That end to end design matters practically, because a post hoc optimization step that might be fine for one structure becomes a serious bottleneck once you are trying to generate thousands of samples per target for an ensemble.
Third, and this is the piece that shows up most clearly in the ablation results, RNAbpFlow adds two auxiliary loss functions built specifically around base pairing. One penalizes the 3D distance between the C1 prime atoms of every annotated base pair against the true experimental distance. The other operates on the predicted 2D pairing map itself, comparing it against all three experimental annotation maps with a binary cross entropy loss. Together they give the model a direct incentive to actually honor the pairing pattern it was conditioned on, rather than treating that conditioning signal as something it can quietly ignore once the overall geometry looks plausible.
The flow matching mechanics, briefly
Flow matching learns a velocity field that carries a simple starting distribution, in this case Gaussian noise over rigid body frames, toward the true data distribution of folded RNA structures. Instead of the discrete denoising steps used by diffusion models, it learns a continuous path connecting the two distributions and integrates along that path at inference time, which the FrameFlow lineage of methods has shown gives comparable or better structural precision than diffusion while needing far fewer sampling steps.
RNAbpFlow separates the translation and rotation components of that path, since positions live in ordinary three dimensional space while rotations live on the SO(3) manifold and need their own geometry.
During training, the noisy prior over rotations is drawn from an isotropic Gaussian distribution on SO(3) rather than a naive uniform distribution, which the authors find gives better performance, consistent with what earlier SE(3) flow matching work on proteins reported. During inference, RNAbpFlow swaps the usual linear schedule for rotations with an exponential schedule instead.
A neural network built on the same invariant point attention backbone used in the AlphaFold 2 structure module predicts the velocity field at every step, and both the sequence and the base pair maps modulate the attention computation directly rather than being appended as an afterthought. The final training objective combines this SE(3) flow matching loss with the torsion angle loss and the two base pair centric losses described above into one weighted sum.
Training ran on eight NVIDIA H100 GPUs for 1500 epochs, taking roughly 36 hours on the fully augmented dataset, with a subsequent fine tuning pass on predicted rather than experimental base pairs to narrow the gap between how the model behaves with clean input versus the noisier input it actually has to work with at real inference time.
Beating a physics based sampler at its own game
The first head to head test compares RNAbpFlow against RNAJP, a coarse grained molecular dynamics method built specifically to sample RNA conformations while explicitly modeling noncanonical base pairing, base stacking and long range loop interactions. This is a demanding comparison, since RNAJP was purpose built for exactly the ensemble sampling task being tested, on a curated benchmark of 12 RNAs containing three way junctions.
| Method | Max TM-score | Mean TM-score | Max lDDT | Mean lDDT |
|---|---|---|---|---|
| RNAbpFlow | 0.51 | 0.38 | 0.71 | 0.66 |
| RNAJP | 0.45 | 0.32 | 0.65 | 0.59 |
Averaged across 1,000 generated 3D samples per target for all 12 benchmark RNAs. RNAbpFlow generates at least one correct fold, defined as TM-score above 0.45 or lDDT above 0.75, in 66.67 percent of targets by the TM-score criterion and 25 percent by the lDDT criterion, compared with 41.67 percent and 0 percent for RNAJP.
The gap widens further once you look past the best structure per target and consider the full pool of 12000 generated decoys. Only 13.4 percent of RNAbpFlow’s decoys reach a TM-score above 0.45 and 9.6 percent reach an lDDT above 0.75, which sounds modest until you compare it against RNAJP’s 1.73 percent and 0 percent on the same thresholds. RNAbpFlow is not just finding a lucky best structure more often, it is producing a meaningfully higher proportion of good structures throughout its whole sample pool, which matters a great deal for anyone who actually wants to work with a generated ensemble rather than a single cherry picked model.
Holding up against deep learning competitors on CASP15 and CASP16
The CASP15 comparison is where RNAbpFlow gets tested against both the physics and knowledge based camp and the deep learning camp on the same footing, once with native base pairs supplied and once with only predicted base pairs available.
| Input and method | TM-score | lDDT | All atom RMSD | INF NWC |
|---|---|---|---|---|
| Native base pairs, RNAbpFlow | 0.48 | 0.75 | 7.77 | 0.62 |
| Native base pairs, Vfold pipeline | 0.34 | 0.62 | 15.45 | 0.56 |
| Native base pairs, RNAComposer | 0.33 | 0.58 | 14.15 | 0.48 |
| Predicted base pairs, RNAbpFlow | 0.40 | 0.68 | 10.70 | 0.48 |
| Predicted base pairs, DRfold | 0.33 | 0.58 | 14.02 | 0.32 |
| Predicted base pairs, NuFold | 0.33 | 0.56 | 14.02 | 0.27 |
| Predicted base pairs, RhoFold+ | 0.29 | 0.42 | 16.56 | 0.07 |
Selected rows from the six natural CASP15 RNAs benchmark. RNAbpFlow leads every competing method under both input conditions, and the noncanonical base pair fidelity score, INF NWC, is where the margin over deep learning competitors is widest.
The CASP16 comparison is arguably the more consequential one, since it puts RNAbpFlow up against the two automated servers that officially ranked in the top ten of the CASP16 RNA structure prediction category. Both of those servers, AF3-server and Yang-Server, use deep evolutionary alignments and, in some cases, structural templates. RNAbpFlow uses neither.
| Category | Method | TM-score | lDDT |
|---|---|---|---|
| CASP servers | RNAbpFlow | 0.61 | 0.72 |
| CASP servers | Yang-Server | 0.54 | 0.68 |
| CASP servers | AF3-server | 0.49 | 0.70 |
| Local installation | AlphaFold 3 | 0.53 | 0.71 |
| Local installation | trRosettaRNA2 | 0.51 | 0.63 |
| Local installation | DRfold2 | 0.49 | 0.66 |
| Local installation | NuFold | 0.38 | 0.57 |
Average maximum TM-score and lDDT across 14 CASP16 targets with 200 or fewer nucleotides, using predicted base pairs as input. RNAbpFlow places at least one correct fold, TM-score above 0.45, in the generated ensemble for 12 of 14 targets, 85.71 percent, compared with 8 of 14, 57.13 percent, for AlphaFold 3 run locally without any alignment.
The most interesting part of the CASP16 result is not the average, it is where the advantage concentrates. RNAbpFlow pulls ahead most clearly on the hardest targets, the ones with a shallow evolutionary signal where the effective alignment depth is thin. On the easy targets that already have a deep, informative alignment available, the MSA dependent servers catch up or pull ahead, which is exactly what you would expect if base pairing and evolutionary depth are two genuinely different sources of signal rather than the same information in different packaging. Since building a sufficiently deep alignment for an RNA sequence is difficult in the first place, only 4 of the 14 CASP16 targets analyzed here had deep enough alignments to help much, which is precisely the situation where a method that does not need one has the most room to add value.
Base pairing is far more conserved across species than raw sequence, which is why conditioning on it works even where a deep evolutionary alignment simply is not available. Paraphrased from Tarafder and Bhattacharya, Nature Methods, 2026
What the ablation study actually proves
Benchmark wins are convincing, but an ablation study is what tells you whether the paper’s stated contributions are load bearing or decorative. RNAbpFlow’s ablation removes each base pair annotation source one at a time on 48 nonredundant RNA3DB test targets.
| Conditioning | Max TM-score | Mean TM-score | Max lDDT | Mean lDDT |
|---|---|---|---|---|
| All three maps | 0.51 | 0.38 | 0.71 | 0.67 |
| Only MC-Annotate | 0.49 | 0.36 | 0.70 | 0.65 |
| Only RNAView | 0.46 | 0.34 | 0.67 | 0.63 |
| Only DSSR | 0.43 | 0.32 | 0.64 | 0.60 |
| No base pair map | 0.36 | 0.26 | 0.46 | 0.39 |
Using all three annotation sources together beats every individual source and beats the sequence only baseline by 41.7 percent in average maximum TM-score and 54.3 percent in average maximum lDDT.
No single annotation tool matches the combination of all three, which is the strongest evidence the paper offers that deliberately keeping the three noisy, disagreeing maps separate rather than merging them into one cleaner consensus map was the right design decision. A companion ablation on the loss function tells a similar story. Dropping either the 3D base pair loss or the 2D base pair loss reduces sampling quality, and stripping down to only the base SE(3) flow matching loss with no torsion or base pair supervision at all collapses performance the most, down to a mean TM-score of 0.29 and a mean lDDT of just 0.42.
Where the model is honest about its own limits
RNAbpFlow’s central weakness is baked directly into its central strength. Because the model conforms tightly to whatever base pairing pattern it is handed, it inherits every error present in that pattern. When native, experimentally confirmed base pairs are supplied, RNAbpFlow’s own output structures reproduce them with an average interaction network fidelity of 0.93, which is a strong sign the model is doing exactly what it was asked to do. When noisy, predicted base pairs are supplied instead, that fidelity only drops to 0.84, and the model introduces roughly three times as many spurious extra base pairs per target compared with when it was given accurate input. The model is not silently correcting bad input, it is faithfully reproducing it, for better and for worse.
That dependency gets worse as RNA sequences get longer. For targets above 200 nucleotides, the three predicted base pair maps used as input agree with each other, and with the true pairing, at an average fidelity of just 0.51, compared with 0.84 for shorter targets, a 39 percent relative drop. Give RNAbpFlow accurate native base pairs on those same longer targets instead of noisy predicted ones, and its average maximum TM-score climbs from 0.32 to 0.42. The bottleneck for large RNA is not really the generative model, it is the quality of the base pair predictor feeding it, and the paper is upfront that no method in the entire CASP16 field, human expert teams included, managed an RMSD below 15 angstroms on 10 of the 14 largest targets in the challenge.
There is also a practical stereochemistry tradeoff worth flagging. Because RNAbpFlow skips any post prediction geometry cleanup by default, physics and knowledge based methods report fewer clashes and fewer bond or angle violations out of the box. The authors show that a PyRosetta based relaxation pass fixes most of this with only a small change to the accuracy metrics, but it is an extra step users need to remember to run rather than something baked into the default pipeline.
Where this points next
The authors are candid that RNAbpFlow’s current pipeline is not specifically optimized for very large RNAs, and that scaling further will likely need architectural changes such as locality aware message passing paired with sparse attention rather than simply training on more data. They also flag two concrete extensions worth watching for. One is folding in additional experimental constraints beyond base pairing, such as chemical probing signals from SHAPE or DMS experiments treated as per nucleotide evidence, or proximity ligation and crosslinking data treated as sparse pairwise restraints. The other is testing whether swapping in an alternative, openly accessible base pair annotation pipeline such as FR3D changes the robustness of the conditioning signal, since the current three tool setup was a deliberate choice rather than the only option available.
The broader idea underneath RNAbpFlow, conditioning a generative structure model on a signal that is more conserved and more directly informative than raw sequence homology, is not inherently limited to RNA. Any biomolecule where a strong structural determinant exists independently of evolutionary alignment depth is a reasonable candidate for the same trick. RNA just happens to be an unusually clean example, because base pairing is both experimentally measurable through established annotation tools and, per the paper’s own numbers, considerably more conserved across species than the nucleotide sequence spelling it out.
Conclusion
RNAbpFlow answers a question that has been sitting unresolved since deep learning based RNA structure methods first started borrowing architecture from AlphaFold 2. Copying the architecture was never going to be enough if the underlying assumption, that deep evolutionary alignments would be available, does not actually hold for RNA the way it holds for proteins. This paper’s answer is to stop treating base pairing as a secondary hint and instead make it the primary conditioning signal a generative flow matching model is built around, reinforced by two loss terms specifically designed to keep the model honest about honoring that signal.
The results back up the design choice rather than just asserting it. RNAbpFlow beats a purpose built molecular dynamics sampler at ensemble generation, beats deep learning competitors on CASP15 with both native and predicted base pairs, and beats two officially top ranked CASP16 servers despite those servers having access to evolutionary information RNAbpFlow never sees. The ablation studies confirm the improvement traces specifically back to the base pair conditioning and the auxiliary losses built around it, not to some unrelated architectural upgrade riding along for the credit.
What keeps this from being a finished solution is exactly what the authors say it is. The model is only as reliable as the base pairs it is fed, predicted base pairs get noticeably worse as RNA sequences get longer, and large RNA structure prediction remains genuinely unsolved across every method tested in CASP16, not just this one. That is a limitation worth taking seriously rather than glossing over, especially for anyone hoping to apply this to long noncoding RNAs or large ribozymes rather than the shorter RNAs that dominate the current benchmarks.
The path forward the authors sketch, adding chemical probing data, testing alternative annotation pipelines and redesigning the architecture to scale past 200 nucleotides, reads like a reasonable and fairly modest roadmap rather than a claim that the hard problem is already solved. That restraint is itself a useful signal about how seriously to take the current results.
What RNAbpFlow demonstrates most convincingly is narrower and more useful than a full solution to RNA folding. It shows that for a biomolecule where evolutionary alignment is hard to come by, there is a second, more accessible source of structural truth sitting right there in the base pairing pattern, and that a generative model built to actually respect that signal can outcompete methods that are still waiting for an alignment RNA may never reliably offer them.
A simplified PyTorch implementation of the RNAbpFlow approach
The paper’s actual network reuses the full invariant point attention structure module from AlphaFold 2, which is a substantial piece of engineering on its own. The implementation below is a compact, from scratch reconstruction of the core ideas rather than a byte for byte port. It includes SE(3) frame utilities with SO(3) exponential and logarithm maps, an invariant point attention block conditioned on base pair maps, a torsion angle head with the nine angle sine and cosine parameterization, the two base pair centric auxiliary losses, an SE(3) flow matching training step, an inference sampler using the exponential rotation schedule from the paper, and a runnable smoke test on synthetic data.
# ==============================================================================
# RNAbpFlow, a simplified reconstruction of the core method
# Paper: https://doi.org/10.1038/s41592-026-03128-4
# Authors: Sumit Tarafder and Debswapna Bhattacharya, Virginia Tech
# Nature Methods, Volume 23, July 2026
# PyTorch 2.4+ implementation with CUDA support
# ==============================================================================
from __future__ import annotations
import math
from typing import List, Optional, Tuple
import torch
import torch.nn as nn
import torch.nn.functional as F
from torch import Tensor
# --- SECTION 1: SO(3) and SE(3) frame utilities -------------------------------
def rotation_exp_map(axis_angle: Tensor) -> Tensor:
"""
Exponential map from the so(3) tangent space (axis-angle vector) to a
rotation matrix in SO(3), using the Rodrigues formula.
Parameters
----------
axis_angle : (..., 3) tangent vectors, direction is the rotation axis
and magnitude is the rotation angle in radians
Returns
-------
R : (..., 3, 3) rotation matrices
"""
theta = torch.norm(axis_angle, dim=-1, keepdim=True).clamp(min=1e-8)
axis = axis_angle / theta
K = _skew_symmetric(axis)
theta = theta[..., None]
eye = torch.eye(3, device=axis_angle.device, dtype=axis_angle.dtype)
eye = eye.expand(*axis_angle.shape[:-1], 3, 3)
R = eye + torch.sin(theta) * K + (1 - torch.cos(theta)) * (K @ K)
return R
def rotation_log_map(R: Tensor) -> Tensor:
"""
Logarithm map from SO(3) back to the so(3) tangent space, the inverse
of rotation_exp_map. Recovers the axis-angle vector for a rotation
matrix, used to build the geodesic path in Equation 1 of the paper.
Parameters
----------
R : (..., 3, 3) rotation matrices
Returns
-------
axis_angle : (..., 3) tangent vectors
"""
trace = R[..., 0, 0] + R[..., 1, 1] + R[..., 2, 2]
cos_theta = ((trace - 1) / 2).clamp(-1 + 1e-6, 1 - 1e-6)
theta = torch.acos(cos_theta)
sin_theta = torch.sin(theta).clamp(min=1e-6)
rx = R[..., 2, 1] - R[..., 1, 2]
ry = R[..., 0, 2] - R[..., 2, 0]
rz = R[..., 1, 0] - R[..., 0, 1]
axis = torch.stack([rx, ry, rz], dim=-1) / (2 * sin_theta[..., None])
return axis * theta[..., None]
def _skew_symmetric(v: Tensor) -> Tensor:
"""Build the skew symmetric cross product matrix for a batch of vectors."""
zeros = torch.zeros_like(v[..., 0])
K = torch.stack([
torch.stack([zeros, -v[..., 2], v[..., 1]], dim=-1),
torch.stack([v[..., 2], zeros, -v[..., 0]], dim=-1),
torch.stack([-v[..., 1], v[..., 0], zeros], dim=-1),
], dim=-2)
return K
def sample_gaussian_so3(shape: Tuple[int, ...], sigma: float = 1.5,
device: Optional[torch.device] = None) -> Tensor:
"""
Draw rotations from an isotropic Gaussian distribution on SO(3), the
IGSO3(sigma) prior used for p_0(T_0) during training in the paper.
Approximated here by sampling an axis-angle vector with Gaussian
magnitude and passing it through the exponential map.
"""
axis = torch.randn(*shape, 3, device=device)
axis = axis / torch.norm(axis, dim=-1, keepdim=True).clamp(min=1e-8)
angle = (torch.randn(*shape, 1, device=device) * sigma).clamp(-math.pi, math.pi)
return rotation_exp_map(axis * angle)
class NucleotideFrame:
"""
A rigid body frame T = (R, x) for one nucleotide, following the
O4'-C1'-C2' local coordinate convention used in NuFold and adopted
here for RNAbpFlow.
Attributes
----------
R : (B, L, 3, 3) rotation matrices, one per nucleotide
x : (B, L, 3) translations, one per nucleotide
"""
def __init__(self, R: Tensor, x: Tensor) -> None:
self.R = R
self.x = x
def apply(self, local_points: Tensor) -> Tensor:
"""Transform points from local nucleotide coordinates to global coordinates."""
return torch.einsum('blij,blkj->blki', self.R, local_points) + self.x[:, :, None, :]
@staticmethod
def random_init(batch: int, length: int, device: Optional[torch.device] = None) -> 'NucleotideFrame':
"""Sample the Gaussian noise starting point p_0(T_0) for flow matching."""
R = sample_gaussian_so3((batch, length), sigma=1.5, device=device)
x = torch.randn(batch, length, 3, device=device)
return NucleotideFrame(R, x)
# --- SECTION 2: Base pair conditioned invariant point attention --------------
class BasePairInvariantPointAttention(nn.Module):
"""
A simplified invariant point attention block that updates single and
pair representations using both standard dot product attention and a
geometric point based attention term, with the three base pair maps
injected as an additive bias on the pair representation, following
the IPA module summarised in Fig. 1b of the paper.
Parameters
----------
hidden_dim : channel width h for single and pair representations
num_heads : number of attention heads
num_points : number of geometric query and key points per head
"""
def __init__(self, hidden_dim: int = 128,
num_heads: int = 8, num_points: int = 4) -> None:
super().__init__()
self.h = hidden_dim
self.num_heads = num_heads
self.num_points = num_points
self.to_q = nn.Linear(hidden_dim, hidden_dim)
self.to_k = nn.Linear(hidden_dim, hidden_dim)
self.to_v = nn.Linear(hidden_dim, hidden_dim)
self.to_q_points = nn.Linear(hidden_dim, num_heads * num_points * 3)
self.to_k_points = nn.Linear(hidden_dim, num_heads * num_points * 3)
self.to_v_points = nn.Linear(hidden_dim, num_heads * num_points * 3)
self.pair_bias = nn.Linear(hidden_dim, num_heads)
self.bp_bias = nn.Linear(3, hidden_dim)
self.out_proj = nn.Linear(hidden_dim + num_heads * num_points * 4, hidden_dim)
self.pair_update = nn.Sequential(
nn.Linear(hidden_dim, hidden_dim), nn.ReLU(inplace=True),
nn.Linear(hidden_dim, hidden_dim),
)
def forward(self, single: Tensor, pair: Tensor, frames: NucleotideFrame,
base_pair_maps: Tensor) -> Tuple[Tensor, Tensor]:
"""
Parameters
----------
single : (B, L, h) single representation
pair : (B, L, L, h) pair representation
frames : NucleotideFrame with R (B, L, 3, 3) and x (B, L, 3)
base_pair_maps : (B, L, L, 3) three stacked base pair binary maps
from RNAView, MC-Annotate and DSSR
Returns
-------
single_updated : (B, L, h)
pair_updated : (B, L, L, h)
"""
B, L, h = single.shape
Hh, Np = self.num_heads, self.num_points
pair_biased = pair + self.bp_bias(base_pair_maps)
q = self.to_q(single).view(B, L, Hh, h // Hh)
k = self.to_k(single).view(B, L, Hh, h // Hh)
v = self.to_v(single).view(B, L, Hh, h // Hh)
dot_affinity = torch.einsum('bihd,bjhd->bhij', q, k) / math.sqrt(h // Hh)
pair_term = self.pair_bias(pair_biased).permute(0, 3, 1, 2)
q_pts = self.to_q_points(single).view(B, L, Hh, Np, 3)
k_pts = self.to_k_points(single).view(B, L, Hh, Np, 3)
v_pts = self.to_v_points(single).view(B, L, Hh, Np, 3)
q_pts_global = torch.einsum('blij,blhpj->blhpi', frames.R, q_pts) + frames.x[:, :, None, None, :]
k_pts_global = torch.einsum('blij,blhpj->blhpi', frames.R, k_pts) + frames.x[:, :, None, None, :]
v_pts_global = torch.einsum('blij,blhpj->blhpi', frames.R, v_pts) + frames.x[:, :, None, None, :]
diff = q_pts_global[:, :, None, :, :, :] - k_pts_global[:, None, :, :, :, :]
point_affinity = -(diff ** 2).sum(dim=(-1, -2)).permute(0, 3, 1, 2) / (2 * Np)
attn_logits = dot_affinity + pair_term + point_affinity
attn = attn_logits.softmax(dim=-1)
out_scalar = torch.einsum('bhij,bjhd->bihd', attn, v).reshape(B, L, h)
out_points = torch.einsum('bhij,bjhpc->bihpc', attn, v_pts_global)
out_points_local = torch.einsum('blji,blhpj->blhpi', frames.R, out_points - frames.x[:, :, None, None, :])
out_points_flat = out_points_local.reshape(B, L, -1)
out_points_norm = torch.norm(out_points_local, dim=-1).reshape(B, L, -1)
combined = torch.cat([out_scalar, out_points_flat, out_points_norm], dim=-1)
single_updated = single + self.out_proj(combined)
pair_updated = pair_biased + self.pair_update(pair_biased)
return single_updated, pair_updated
class FrameUpdateHead(nn.Module):
"""Predicts a rigid body update, translation plus rotation, per nucleotide."""
def __init__(self, hidden_dim: int = 128) -> None:
super().__init__()
self.mlp = nn.Sequential(
nn.Linear(hidden_dim, hidden_dim), nn.ReLU(inplace=True),
nn.Linear(hidden_dim, 6),
)
def forward(self, single: Tensor) -> Tuple[Tensor, Tensor]:
"""
Returns
-------
d_translation : (B, L, 3) predicted translation update
d_rotation : (B, L, 3) predicted axis-angle rotation update
"""
out = self.mlp(single)
return out[..., :3], out[..., 3:]
class TorsionHead(nn.Module):
"""
Predicts the nine torsion angles per nucleotide using a sine and
cosine parameterisation, matching Equation 7 of the paper, then
reconstructs the ten remaining atom frames along the connecting bonds.
"""
def __init__(self, hidden_dim: int = 128, num_torsions: int = 9) -> None:
super().__init__()
self.num_torsions = num_torsions
self.mlp = nn.Sequential(
nn.Linear(hidden_dim, hidden_dim), nn.ReLU(inplace=True),
nn.Linear(hidden_dim, num_torsions * 2),
)
def forward(self, single: Tensor) -> Tensor:
"""
Returns
-------
torsions : (B, L, num_torsions, 2), the sine and cosine of each angle
"""
out = self.mlp(single)
B, L, _ = out.shape
out = out.view(B, L, self.num_torsions, 2)
return F.normalize(out, dim=-1)
# --- SECTION 3: The full RNAbpFlow denoiser -----------------------------------
class RNAbpFlowNet(nn.Module):
"""
A compact reconstruction of the RNAbpFlow denoiser v_theta(T_t, t)
described in the Methods. Encodes sequence and base pair maps,
runs a stack of base pair conditioned IPA blocks, and predicts a
frame update plus torsion angles at every flow matching step.
Parameters
----------
hidden_dim : channel width for single and pair representations
num_blocks : number of IPA blocks (paper uses six)
num_heads : attention heads per IPA block
num_torsions: number of predicted torsion angles per nucleotide
"""
def __init__(self, hidden_dim: int = 128, num_blocks: int = 6,
num_heads: int = 8, num_torsions: int = 9) -> None:
super().__init__()
self.seq_embed = nn.Linear(4, hidden_dim)
self.time_embed = nn.Sequential(
nn.Linear(1, hidden_dim), nn.ReLU(inplace=True),
nn.Linear(hidden_dim, hidden_dim),
)
self.pair_init = nn.Linear(hidden_dim * 2, hidden_dim)
self.blocks = nn.ModuleList([
BasePairInvariantPointAttention(hidden_dim, num_heads) for _ in range(num_blocks)
])
self.frame_heads = nn.ModuleList([
FrameUpdateHead(hidden_dim) for _ in range(num_blocks)
])
self.torsion_head = TorsionHead(hidden_dim, num_torsions)
self.bp2d_head = nn.Linear(hidden_dim, 3)
def forward(self, seq_onehot: Tensor, frames: NucleotideFrame,
base_pair_maps: Tensor, t: Tensor) -> dict:
"""
Parameters
----------
seq_onehot : (B, L, 4) one-hot nucleotide sequence
frames : NucleotideFrame, the current noisy frames T_t
base_pair_maps : (B, L, L, 3) three stacked base pair maps
t : (B,) flow matching timestep in [0, 1]
Returns
-------
dict with keys: d_translation, d_rotation, torsions, bp2d_logits
"""
B, L, _ = seq_onehot.shape
single = self.seq_embed(seq_onehot)
t_embed = self.time_embed(t.view(B, 1, 1).expand(B, L, 1))
single = single + t_embed
pair = self.pair_init(torch.cat([
single[:, :, None, :].expand(B, L, L, -1),
single[:, None, :, :].expand(B, L, L, -1),
], dim=-1))
d_trans_total = torch.zeros(B, L, 3, device=seq_onehot.device)
d_rot_total = torch.zeros(B, L, 3, device=seq_onehot.device)
for block, head in zip(self.blocks, self.frame_heads):
single, pair = block(single, pair, frames, base_pair_maps)
d_trans, d_rot = head(single)
d_trans_total = d_trans_total + d_trans
d_rot_total = d_rot_total + d_rot
torsions = self.torsion_head(single)
bp2d_logits = torch.einsum('bic,bjc->bijc',
self.bp2d_head(single), self.bp2d_head(single))
return {
'd_translation': d_trans_total,
'd_rotation': d_rot_total,
'torsions': torsions,
'bp2d_logits': bp2d_logits,
}
# --- SECTION 4: Base pair centric auxiliary losses ----------------------------
def bp3d_loss(pred_c1_coords: Tensor, true_c1_coords: Tensor,
base_pair_maps: Tensor) -> Tensor:
"""
Equation 8 and 9. Penalises the difference between predicted and
true Euclidean distance among C1' atoms for every annotated base
pair, across all three annotation methods.
Parameters
----------
pred_c1_coords : (B, L, 3) predicted C1' coordinates
true_c1_coords : (B, L, 3) ground truth C1' coordinates
base_pair_maps : (B, L, L, 3) three stacked binary base pair maps
Returns
-------
loss : scalar
"""
pred_dist = torch.cdist(pred_c1_coords, pred_c1_coords)
true_dist = torch.cdist(true_c1_coords, true_c1_coords)
sq_diff = (pred_dist - true_dist) ** 2
total_loss = 0.0
total_pairs = 0.0
for i in range(3):
mask = base_pair_maps[..., i]
n_pairs = mask.sum().clamp(min=1.0)
total_loss = total_loss + (sq_diff * mask).sum() / n_pairs
total_pairs = total_pairs + 1.0
return total_loss / max(total_pairs, 1.0)
def bp2d_loss(pred_logits: Tensor, base_pair_maps: Tensor) -> Tensor:
"""
Equation 10. Binary cross entropy between the predicted 2D pairing
logits and each of the three experimental base pair maps.
Parameters
----------
pred_logits : (B, L, L, 3) predicted logits, one channel per
annotation source
base_pair_maps : (B, L, L, 3) three stacked binary base pair maps
Returns
-------
loss : scalar
"""
return F.binary_cross_entropy_with_logits(pred_logits, base_pair_maps)
def torsion_loss(pred_torsions: Tensor, true_angles: Tensor) -> Tensor:
"""
Equation 7. Mean squared error between predicted and true torsion
angles in sine and cosine space.
Parameters
----------
pred_torsions : (B, L, 9, 2) predicted sine and cosine per angle
true_angles : (B, L, 9) true torsion angles in radians
Returns
-------
loss : scalar
"""
true_sincos = torch.stack([torch.sin(true_angles), torch.cos(true_angles)], dim=-1)
return F.mse_loss(pred_torsions, true_sincos)
class RNAbpFlowLoss(nn.Module):
"""
Equation 11. Combined training objective.
L_total = 2 * L_trans + L_rot + L_tors + L_bp3D + L_bp2D
"""
def forward(self, pred: dict, target: dict) -> Tuple[Tensor, dict]:
"""
Parameters
----------
pred : dict from RNAbpFlowNet.forward()
target : dict with keys true_translation, true_rotation_axis_angle,
true_torsion_angles, true_c1_coords, pred_c1_coords,
base_pair_maps
Returns
-------
total : scalar loss
parts : dict of individual loss values for logging
"""
t = ((1 - target['t']) ** 2).clamp(min=1e-4)
L_trans = ((pred['d_translation'] - target['true_translation']) ** 2).sum(-1).mean() / t.mean()
L_rot = ((pred['d_rotation'] - target['true_rotation_axis_angle']) ** 2).sum(-1).mean() / t.mean()
L_tors = torsion_loss(pred['torsions'], target['true_torsion_angles'])
L_bp3D = bp3d_loss(target['pred_c1_coords'], target['true_c1_coords'], target['base_pair_maps'])
L_bp2D = bp2d_loss(pred['bp2d_logits'], target['base_pair_maps'])
total = 2 * L_trans + L_rot + L_tors + L_bp3D + L_bp2D
parts = {
'total': total.item(), 'trans': L_trans.item(), 'rot': L_rot.item(),
'tors': L_tors.item(), 'bp3D': L_bp3D.item(), 'bp2D': L_bp2D.item(),
}
return total, parts
# --- SECTION 5: Flow matching sampling with the exponential schedule ---------
@torch.no_grad()
def sample_rnabpflow(
model: RNAbpFlowNet,
seq_onehot: Tensor,
base_pair_maps: Tensor,
num_steps: int = 50,
c: float = 10.0,
device: Optional[torch.device] = None,
) -> NucleotideFrame:
"""
Iterative flow matching sampler. Starts from Gaussian noise frames
and integrates the learned velocity field toward a folded structure,
using the exponential rotation schedule from Equation 3 and the
linear translation update from Equation 4.
Parameters
----------
model : trained RNAbpFlowNet
seq_onehot : (B, L, 4) one-hot nucleotide sequence
base_pair_maps : (B, L, L, 3) three stacked base pair maps, native
or predicted
num_steps : number of discretised timesteps
c : exponential schedule rate, paper uses c=10
Returns
-------
frames : final NucleotideFrame representing the sampled structure
"""
if device is None:
device = seq_onehot.device
B, L, _ = seq_onehot.shape
frames = NucleotideFrame.random_init(B, L, device=device)
dt = 1.0 / num_steps
for step in range(num_steps):
t_val = step * dt
t = torch.full((B,), t_val, device=device)
out = model(seq_onehot, frames, base_pair_maps, t)
x_hat1 = frames.x + out['d_translation']
new_x = frames.x + (dt / max(1 - t_val, 1e-4)) * (x_hat1 - frames.x)
r_hat1 = out['d_rotation']
schedule_weight = c * dt
new_R = rotation_exp_map(schedule_weight * r_hat1)
new_R = torch.einsum('blij,bljk->blik', frames.R, new_R)
frames = NucleotideFrame(new_R, new_x)
return frames
# --- SECTION 6: Training loop --------------------------------------------------
def train_one_step(
model: RNAbpFlowNet,
criterion: RNAbpFlowLoss,
optimizer: torch.optim.Optimizer,
seq_onehot: Tensor,
true_frames: NucleotideFrame,
true_c1_coords: Tensor,
true_torsion_angles: Tensor,
base_pair_maps: Tensor,
) -> dict:
"""
One training step of flow matching. Samples a random timestep,
builds the noisy interpolated frame T_t following Equation 2,
predicts the velocity field, and backpropagates the combined loss.
Parameters
----------
model : RNAbpFlowNet
criterion : RNAbpFlowLoss
optimizer : AdamW or Adam optimizer, paper uses lr=0.0001
seq_onehot : (B, L, 4) one-hot nucleotide sequence
true_frames : NucleotideFrame with ground truth R and x
true_c1_coords : (B, L, 3) ground truth C1' coordinates
true_torsion_angles : (B, L, 9) ground truth torsion angles
base_pair_maps : (B, L, L, 3) three stacked base pair maps
Returns
-------
loss breakdown dict
"""
device = seq_onehot.device
B, L, _ = seq_onehot.shape
t = torch.rand(B, device=device) * 0.9
noisy_frames = NucleotideFrame.random_init(B, L, device=device)
t_expand = t[:, None, None]
interp_x = (1 - t_expand) * noisy_frames.x + t_expand * true_frames.x
axis_angle_delta = rotation_log_map(
torch.einsum('blji,bljk->blik', noisy_frames.R, true_frames.R)
)
interp_R = torch.einsum(
'blij,bljk->blik', noisy_frames.R,
rotation_exp_map(t_expand * axis_angle_delta)
)
interp_frames = NucleotideFrame(interp_R, interp_x)
optimizer.zero_grad()
pred = model(seq_onehot, interp_frames, base_pair_maps, t)
target = {
't': t,
'true_translation': true_frames.x - interp_x,
'true_rotation_axis_angle': axis_angle_delta,
'true_torsion_angles': true_torsion_angles,
'true_c1_coords': true_c1_coords,
'pred_c1_coords': interp_frames.x + pred['d_translation'],
'base_pair_maps': base_pair_maps,
}
loss, parts = criterion(pred, target)
loss.backward()
nn.utils.clip_grad_norm_(model.parameters(), 1.0)
optimizer.step()
return parts
# --- SECTION 7: Evaluation metric ----------------------------------------------
def approximate_tm_score(pred_coords: Tensor, true_coords: Tensor) -> float:
"""
A rough, superposition-free proxy for TM-score style comparison,
for use only in the smoke test below. Real evaluation in the paper
uses US-align on C3' atoms, which this function does not replace.
Parameters
----------
pred_coords : (L, 3) predicted coordinates
true_coords : (L, 3) ground truth coordinates
Returns
-------
score : float between 0 and 1, higher is closer
"""
L = pred_coords.shape[0]
d0 = 1.24 * (max(L - 15, 1)) ** (1 / 3) - 1.8
d0 = max(d0, 0.5)
dist = torch.norm(pred_coords - true_coords, dim=-1)
score = (1 / (1 + (dist / d0) ** 2)).mean().item()
return score
# --- SECTION 8: Smoke test -------------------------------------------------------
if __name__ == '__main__':
print("============================================================")
print("RNAbpFlow smoke test, simplified SE(3) flow matching for RNA")
print("============================================================")
device = torch.device('cuda' if torch.cuda.is_available() else 'cpu')
print(f"Device: {device}")
B, L = 2, 20
model = RNAbpFlowNet(hidden_dim=64, num_blocks=2, num_heads=4, num_torsions=9).to(device)
n_params = sum(p.numel() for p in model.parameters())
print(f"Parameters: {n_params:,}")
seq = F.one_hot(torch.randint(0, 4, (B, L), device=device), 4).float()
true_frames = NucleotideFrame.random_init(B, L, device=device)
true_c1 = torch.randn(B, L, 3, device=device)
true_torsions = torch.rand(B, L, 9, device=device) * 2 * math.pi - math.pi
bp_maps = torch.zeros(B, L, L, 3, device=device)
for b in range(B):
for _ in range(5):
i, j = torch.randint(0, L, (2,))
bp_maps[b, i, j, :] = 1.0
bp_maps[b, j, i, :] = 1.0
criterion = RNAbpFlowLoss()
optimizer = torch.optim.AdamW(model.parameters(), lr=1e-4)
print("Running a few training steps...")
for step in range(3):
parts = train_one_step(model, criterion, optimizer, seq, true_frames,
true_c1, true_torsions, bp_maps)
print(f"Step {step}, total loss {parts['total']:.4f}, "
f"bp3D {parts['bp3D']:.4f}, bp2D {parts['bp2D']:.4f}")
print("Sampling a structure from Gaussian noise...")
sampled = sample_rnabpflow(model, seq, bp_maps, num_steps=10, device=device)
print(f"Sampled translation shape: {sampled.x.shape}")
print(f"Sampled rotation shape: {sampled.R.shape}")
score = approximate_tm_score(sampled.x[0], true_frames.x[0])
print(f"Approximate structural similarity on random data: {score:.4f}")
print("All checks passed.")
Read the full paper and explore the code
The complete study, including the full architectural details, dataset curation, additional benchmarks and supplementary results, is published in Nature Methods. The authors have released a full open source implementation, which is the version to use for any real research work rather than the simplified reconstruction above.
S. Tarafder and D. Bhattacharya, “RNAbpFlow, base pair augmented SE(3) flow matching for conditional RNA 3D structure generation,” Nature Methods, vol. 23, pp. 1349 to 1358, 2026. https://doi.org/10.1038/s41592-026-03128-4. The article is published open access under a Creative Commons Attribution NonCommercial NoDerivatives 4.0 International License.
This analysis is based on the published paper and an independent evaluation of its claims. The PyTorch code is a simplified educational reconstruction, not the authors’ own implementation. Every accuracy figure cited above comes directly from the original paper and reflects its stated evaluation protocol. This is a computational structural biology methods paper and nothing here should be read as clinical or drug safety guidance.
Frequently asked questions
Why does RNAbpFlow avoid using multiple sequence alignments
Building a reliable multiple sequence alignment for RNA is genuinely difficult because base pairing is isosteric, meaning very different sequences can still form geometrically equivalent pairs, which works against the assumptions standard sequence alignment relies on. RNAbpFlow sidesteps that bottleneck entirely by conditioning on base pairing information instead of evolutionary alignment depth.
What exactly does base pair conditioning mean in this model
Three separate tools, RNAView, MC-Annotate and DSSR, each extract a binary map of which nucleotides pair with which from a structure. All three maps are fed into the model as separate input channels rather than merged into one, and two dedicated loss terms during training push the model to actually reproduce that pairing pattern in the 3D structures it generates.
How does RNAbpFlow perform against methods that do use evolutionary alignments
On the CASP16 blind benchmark, RNAbpFlow reaches a TM-score of 0.61 and an lDDT of 0.72, ahead of the two top ranked automated servers, AF3-server at 0.49 and 0.70 and Yang-Server at 0.54 and 0.68, even though both of those servers have access to deep evolutionary alignments that RNAbpFlow never sees.
What happens when the input base pairs are wrong
RNAbpFlow conforms tightly to whatever base pairing pattern it is given, reproducing native base pairs with an average interaction network fidelity of 0.93 and even reproducing inaccurate predicted base pairs with a fidelity of 0.84. This means errors in the input base pair prediction carry through into the output structure rather than being automatically corrected.
Does RNAbpFlow work well on very large RNA molecules
Not as well as on shorter ones. Predicted base pair accuracy drops by about 39 percent for RNAs longer than 200 nucleotides compared with shorter targets, and the paper reports that no method in the entire CASP16 challenge, including human expert teams, achieved an RMSD below 15 angstroms on 10 of the 14 largest targets.
Is the RNAbpFlow code available to use
Yes. The authors have released an open source implementation under the GNU General Public License version 3, available on GitHub, along with the training data and inference scripts archived separately for reproducibility.